You must be signed in to read the rest of this article.
Registration on CDEWorld is free. Sign up today!
Forgot your password? Click Here!
The American Dental Association (ADA)1 and many specialty groups in dentistry—and medicine for that matter—have developed and published monitoring guidelines for their members. In terms of respiratory monitoring, these guidelines are similar in most regards, and they all adhere to those suggested currently for non-anesthesiologists by the American Society of Anesthesiology (ASA).2 Principles of respiratory monitoring were presented in a previous continuing education article in this journal.3
The focus of this article is cardiovascular monitoring. General anesthesia has significant influences on the heart and blood pressure, but there is general agreement that moderate and even deep sedation have minimal impact on cardiovascular function.2 The rationale for cardiovascular monitoring during sedation stems from the realization that sedation may blunt appropriate autonomic responses associated with procedural stresses. Furthermore, if sedation is inadequate, the patient may respond with potentially detrimental autonomic responses, such as tachycardia and hypertension. Early detection of these cardiovascular changes may lead the practitioner to intervene earlier and thus reduce the risk of complications from these changes. Published guidelines strongly agree that monitoring heart rate and blood pressure decrease the likelihood of adverse outcomes related to sedation during dental procedures. However, there are differences in opinion regarding the frequency with which these vital signs should be recorded and the necessity for continuous electrocardiographic (ECG) monitoring during moderate sedation. These issues will be addressed within appropriate sections of this article.
Blood Pressure: Physiological Considerations
Despite the complexity of cardiovascular physiology, the essential purpose of this system is rather simplistic; it must perfuse vital organs and tissues with oxygenated blood. The heart serving as the pump is pivotal for this process. The cardiac cycle comprises all events that occur from the beginning of one ventricular systole until the beginning of a second. The ventricles fill with blood during diastole (relaxation) and eject blood into the pulmonary artery and aorta during systole (contraction). The volume ejected into the aorta during each cycle is called stroke volume, and the total volume ejected each minute (rate X stroke volume) is the cardiac output (Figure 1).
In any closed system composed of a pump and tubing, pressure is proportional to the amount of fluid pumped and the amount of resistance imparted by the tubing. In the cardiovascular system this translates to: blood pressure = cardiac output X arterial resistance. Therefore, blood pressure increases when there is an increase in cardiac output, from either an increase in stroke volume or heart rate. Likewise, blood pressure will also increase with an increase in arterial resistance caused by a decrease in diameter of the arteries.
Blood pressure fluctuates continuously due to the cyclic nature of the pumping action of the heart. The highest pressure occurs during ventricular contraction (systole) and is designated systolic blood pressure (SBP). The lowest pressure occurs during ventricular relaxation (diastole) and is, therefore, designated as diastolic blood pressure (DBP). Mean arterial pressure (MAP) is the time-weighted average of the blood pressure throughout the cardiac cycle. It is not a true average of systolic and diastolic pressures because greater time is spent in diastole; systole occupies ~1/3 and diastole ~2/3 of a cycle. Therefore, MAP is conventionally estimated by the following formula4:
| MAP = | SBP + 2(DBP) |
| 3 |
Systolic blood pressure is the result of force provided by ventricular contraction or systole. Cardiac output (minute output of the heart) sustains this pressure and, therefore, it can be influenced by heart rate and stroke volume. Of these two factors, stroke volume is most significant because it provides the “surge” that creates the systolic pressure. Rate acts merely as a “compensator” for changes in stroke volume. For example, slow rates are common in well-trained athletes because of conditioning. They have increased stroke volumes at rest and require slower heart rates to maintain an adequate cardiac output. Patients in heart failure have lower stroke volumes and require a faster heart rate to maintain the same cardiac output.
At the completion of systole, the ventricles enter a period of relaxation (diastole), and their intraventricular pressure approaches zero. However, blood (aortic) pressure does not decline this far because resistance within the arterial system sustains a diastolic pressure (Figure 2). The factor responsible for diastolic pressure is variably called aortic resistance, systemic vascular resistance, or arterial resistance. Although blood volume and viscosity are contributory, arterial diameter is the principal determinant of this resistance. Therefore, drugs that constrict arteries will increase diastolic pressure primarily, but systolic pressure will also increase as the force of ventricular contraction must increase to eject against this resistance. Conversely, drugs that produce arterial dilation will decrease diastolic and systolic pressure.
Stroke volume is the principal determinant of systolic pressure and factors that influence its intensity must be appreciated. Myocardial cells have an inherent degree of strength called contractility. This force of contraction is reflected as a so-called “ejection fraction,” which represents the actual portion or fraction of the end-diastolic volume that is ejected as stroke volume. A heart having an ejection fraction of 70% has greater contractility than one having an ejection fraction of 40%. Sympathetic stimulation of myocardial cells increases contractility and subsequent stroke volume.
Venous return to the heart exerts a tension or “stretch” on the heart muscle referred to as “preload.” According to the Frank-Starling principle, striated muscle contracts more forcefully as it is stretched. An increase in venous return will increase end-diastolic volume (preload), and this results in a greater stroke volume. This principle has a limit, however. At a certain point, the heart will become so stretched and strained that subsequent stroke volumes will decrease. This principle is illustrated in Figure 3 where a normal heart is compared with one that is in failure.
At the beginning of systole, the myocardial wall develops tension. It is influenced dramatically by any resistance the ventricles must overcome to eject its contents. This resistance is called “afterload.” The most obvious factor that influences afterload is aortic or arterial resistance, reflected as diastolic blood pressure. A higher diastolic pressure will increase the afterload on myocardial fibers attempting to open the aortic valve to eject their stroke volume. Although imprecise, it is common practice to use the terms afterload and arterial resistance as synonyms. An increase in afterload has a negative influence on stroke volume, but a healthy heart is able to adapt and continue to eject a consistent stroke volume up to a point. However, a point is reached where stroke volume will commence to decline. This happens at lower values if a heart is compromised. A flowchart illustrating the determinants of arterial pressure is presented in Figure 4.
Blood Pressure: Monitoring Considerations
The most accurate measurement of arterial blood pressure is by direct monitoring. This involves the placement of a catheter directly into an artery (arterial line) and connecting it to an electronic device that records beat-to-beat pressure readings as well as a pulsatile waveform. The indications for so-called invasive monitoring are for critically ill patients where wide swings in blood pressure or fluid shifts are anticipated, and blood pressure must be monitored on a minute-to-minute basis. This, of course, is never the case in outpatient practice where blood pressure is reasonably approximated using indirect methods.
The indirect method for recording blood pressure was developed by employing auscultation using a stethoscope and a sphygmomanometer attached to a column of mercury, the height of which reflects the blood pressure. This accounts for the conventional reporting of blood pressure in millimeters of mercury (mm Hg) or torr. The mercury manometer is considered the “gold standard” because it gives an accurate result and does not required periodic calibration. Today, auscultation methods using aneroid manometers or oscillometric electronic devices are used more routinely for patient monitoring.
An inflatable cuff is placed around the upper arm at roughly the same height as the heart and is attached to either a manometer or electronic device. The cuff is inflated until blood flow is occluded and then is slowly deflated. A stethoscope is placed below the cuff at the elbow in the area of the brachial artery. As blood starts to flow through the brachial artery there is turbulence, and it creates the first of a series of Korotkoff sounds. The reading at which this sound is first heard is designated the systolic blood pressure. As the cuff continues to be deflated, the sounds first become louder, then duller and finally muffled. A point is reached where blood flow is no longer impeded and turbulence subsides. At this point, the Korotkoff sounds disappear, signaling the diastolic blood pressure. While the absence of sound coincides with diastolic pressure in most patients, diastolic pressure in children is more accurately recorded when the sound becomes muffled.5
Palpation of arterial pulses can also be used to estimate systolic blood pressure, most often in emergency situations. Minimum blood pressures to generate a palpable arterial pulse in designated locations include radial (80 mm Hg), femoral (70 mm Hg), and carotid (60 mm Hg). Also, a reasonably accurate systolic pressure can be quickly ascertained by inflating the cuff and deflating it until a radial pulse is detected.
Both palpation and auscultatory methods are prone to errors as sound and tactile sensations are subjective and may not be identified properly. Palpated pressure is generally 2 mm to 5 mm Hg lower than that measured by auscultation.5 Cuff deflation that is too rapid can lead to underestimation of blood pressure and improper cuff size is another source for error. To obtain accurate measurements, it is recommended that the cuff width be approximately 20% to 50% greater than the diameter of the extremity being used.4 The bladder length should also be sufficient to encircle at least 50% to 60% of the extremity. Falsely high estimates may occur if too small a cuff is used or if it is applied too loosely because greater pressure will be required to occlude the artery. Falsely low blood pressures will be recorded if too large a cuff is used because less pressure is required to occlude the artery. Ideally, the cuff should be placed on the upper arm at the height of the heart to avoid errors due to gravity. Cuff placement above the heart will provide lower pressure readings and placement below the heart will provide higher readings.
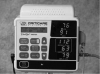
Automated microprocessor-controlled oscillometric monitors have largely replaced the palpation and auscultatory methods of blood pressure monitoring. These noninvasive blood pressure monitors make use of a pressure transducer that measures cuff pressure and processes it into a digital number. The cuff inflates automatically, maintaining a constant pressure while sampling for oscillations. If no oscillations are sensed, then a deflation valve is opened and the next level is sampled for oscillations indicating blood flow is present. Current oscillometric units are able to record the pulse rate along with systolic, diastolic, and mean arterial blood pressures at intervals defined by the operator. Many of these units include pulse oximetry measurement as well (Figure 5). Noninvasive blood pressure units are ideal for patient monitoring during sedation. They require less skill to use than manual devices and free the person responsible for monitoring to do other patient-related tasks.
As stated previously, guidelines for cardiovascular monitoring are not as well defined as those for respiratory monitoring. All guidelines, including those proposed by the ASA,2 are based on opinion rather than published data. Cardiovascular function is usually maintained or unaffected throughout the continuum of sedation, and information and publications are insufficient to support firm conclusions. Nevertheless, consultants strongly agree that regular monitoring of blood pressure decreases the likelihood of adverse outcomes related to sedation during dental procedures. The frequency at which blood pressure should be determined and recorded is still debated. Guidelines for blood pressure monitoring during general anesthesia require assessment and recordings every 5 minutes at a minimum, but recommendations during sedation typically range from this interval to every 30 minutes. The frequency of monitoring and recording the blood pressure should be determined by the dentist based on the perceived needs of the individual patient. Given the minimal impact of all sedation levels on cardiovascular function and the general lack of consensus among published guidelines, we believe the suggestions summarized in the Table are reasonable for assessment and recording in the sedation record.
Heart Rate and Rhythm: Physiological Considerations
An adequate cardiac output must be sustained to provide sufficient blood flow throughout the body. Heart rate and rhythm are critical in this regard and the regulation of these electrophysiologic events must be appreciated. The heart is composed of two primary types of cells: those comprising the working myocardium and the conduction system cells. The myocardial cells have the ability to contract when electrically stimulated. The cells of the conduction system provide this stimulus by transmitting impulses (action potentials) throughout the myocardium to achieve a coordinated rhythm of contraction and relaxation among and between the atria and ventricles.
The normal sequence of action potentials originates in the sinoatrial (SA) node and is conducted to its endpoint in the ventricular myocardium along specialized tissue pathways (Figure 6). From the SA node, the impulse travels to the atrioventricular (AV) node by discrete internodal pathways. From the AV node, conduction travels by a pathway known as the Bundle of His, which divides into right and left bundles. The terminal portions of these bundles are the Purkinje fibers, which are responsible for the direct stimulation of the ventricular myocardial cells.
The SA node is regarded as the pacemaker because it has the highest spontaneous rate of depolarization triggering an action potential. It has a resting frequency of 70 to 80 per minute and will override the spontaneous rates of the AV node and Purkinje system. Should the SA node fail, these other areas of the conduction system will act as backup pacemakers. The AV node has an intrinsic rate of 40 to 60 per minute and the Purkinje system a rate of 15 to 30 per minute.6 Impulses that originate from these locations are sometimes referred to as escape beats.
Though the SA node has an intrinsic rate of 70 to 80 per minute, autonomic nervous system activity can modulate this rate. Stimulation by the sympathetic nervous system and release of circulating catecholamines will increase the heart rate by virtue of beta-1 receptor stimulation within the SA node. Conversely, vagal activity through the parasympathetic nervous system will slow the heart rate by activating cholinergic (muscarinic) receptors. Both divisions exert similar influences on the AV node but only sympathetic innervation is present within the Bundle of His and Purkinje fibers.
Autonomic control of heart rate will respond to demands placed on the patient and may be initiated via several baroreceptor-mediated reflexes. Arterial baroreceptors in the carotid sinus and aortic arch react to alterations in stretch of these vessels by changes in blood pressure. When stretched by an increase in blood pressure, sympathetic outflow is inhibited, leading to vasodilation, while a relative increase in vagal tone decreases heart rate. Collectively, this results in a decrease in blood pressure. Likewise, stretch receptors, similar to baroreceptors, are located in the right atrium and react to stretch from increased filling pressures. This is referred to as the Bainbridge reflex and increases heart rate in an effort to accommodate any sudden increase in venous return.
Should the body require greater cardiac output, the heart rate will generally be increased. However, extremes of heart rate may be detrimental to cardiac output and blood pressure. Based on the formula CO = HR X SV, changes in heart rate and/or stroke volume will affect cardiac output. The ability of the heart to increase cardiac output is limited, however, and after a certain heart rate we see a leveling off or an actual decrease in stroke volume. This is because filling of the left ventricle takes place during diastole or the resting/relaxation phase of the cardiac cycle. With increases in heart rate, the time in diastole is shortened, and there is less time for filling. Further increase in heart rate may actually result in a decline in blood pressure. The precise rate at which ventricular filling declines is highly variable and depends on the size of the ventricular chambers and the compliance or ‘‘stiffness’’ of the myocardial wall. In the healthy patient, heart rates up to ~120 beats per minute will generally allow adequate filling. On the other end of the spectrum, a slower heart rate (eg, <50 beats per minute) may allow for more time in diastole, but ventricular filling becomes maximized. At this point, stroke volume cannot increase further to compensate and any further decrease in heart rate will lead to a decrease in cardiac output and blood pressure. Again, the actual heart rate at which cardiac output will decline is highly variable depending on the patient’s cardiac status.
A mild tachycardia (HR > 100) may lead to an increase in cardiac output, but it can be detrimental to a patient with coronary artery disease (CAD). The hallmark of CAD is a narrowing of coronary arteries limiting supply of oxygen to both myocardial contractile and neural conductive cells. Patients with CAD are always at risk for ischemic events, and prevention is dependent on maintaining a favorable balance between myocardial oxygen supply and demand. Perfusion of the myocardium occurs primarily during diastole and is driven by the gradient between aortic diastolic pressure and ventricular end-diastolic pressure. When heart rate increases, the diastolic period is shortened and may reduce time for coronary perfusion. Furthermore, a greater heart rate increases the heart’s requirement for oxygen. Therefore, rapid heart rates not only reduce supply but increase oxygen demand. This accounts for the pivotal role of beta-blockers in the medical management of coronary artery disease.
There has been a long-standing interest in developing a method to assess myocardial oxygen demand in the clinical setting. Two indices have been developed: (a) rate-pressure product (HR X MAP) and (b) pressure/rate quotient (MAP/HR). Neither method has been found precisely accurate in predicting myocardial ischemia.7 Both indices fail to address the impact of low pressures that provide inadequate coronary perfusion. A rate-pressure product can be normal or low, but the patient may still be at risk for ischemia especially in a situation where the heart rate is fast and the blood pressure is low. Again, this creates a huge imbalance between myocardial oxygen supply and demand. Simply stated, when managing patients with coronary artery disease we need to avoid rapid heart rates and extremes in blood pressure, high or low.
Heart Rate and Rhythm: Monitoring Considerations
Monitoring of heart rate can be accomplished by many different methods, with direct palpation of a radial or carotid pulse most obvious. The pulse oximeter monitors adequacy of oxygenation for patients undergoing sedation but also provides useful information pertaining to heart rate. Because the pulse oximeter requires pulsatile blood flow, the character of its tone with each beat allows the practitioner to assess oxygen saturation as well as changes in heart rate or rhythm without looking directly at the monitor. Pulse oximeter readings are subject to interference from electrical activity such as cautery units or if certain low flow states exist in the extremity being used for monitoring. Technical issues regarding the use of pulse oximeters were presented more thoroughly in a previous article.3
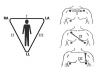
The ECG is also a useful monitor for heart rate, and a better assessment of rhythm. Conventional ECG monitoring uses a three-lead system, commonly known as limb leads based on the familiar Einthoven’s triangle (Figure 7). More sophisticated ECGs can monitor up to 12 leads and provide extensive information regarding ischemia, hypertrophy, and dysrhythmias. However, a three-lead device is adequate for monitoring heart rate and rhythms during sedation. Generally, lead two tracings provide the largest complexes, especially the P wave, which allows the best assessment of rhythm. While the use of electrodes will allow inspection of any of the three leads, wrist clips can only provide a lead one tracing.
A key point is that an ECG monitors only electrical activity. It does not assure the complexes are generating myocardial contraction; they could merely represent pulseless electrical activity. For this reason, the pulse of an unresponsive patient must be verified by palpation. While the pulse oximeter and ECG both monitor heart rate, it is the pulse oximeter that assures pulsatile blood flow and is most useful for monitoring pulse. The ECG supplements this information and adds an ability to detect rhythm disturbances. Technical issues regarding electrocardiography and rhythm interpretation were presented more thoroughly in a previous continuing education article.8
Published guidelines for patient monitoring during sedation are consistent in requiring the continuous assessment of oxygenation by pulse oximetry. Therefore, continuous monitoring of heart rate is also provided. Issues regarding requirements for ECG monitoring are less consistent. All agree that sedation has minimal impact on cardiovascular function, but the ASA guidelines nevertheless require ECG monitoring for even moderate levels of sedation.2 There is no data assessment of scientific outcomes for the basis of this recommendation, but from their perspective it is understandable. All monitoring systems used by anesthesiologists include ECG and any case scheduled initially for sedation may require instant conversion to a full general anesthetic. However, this may be excessive for the dentist providing moderate sedation, and guidelines published by the ADA are more practical.1 These guidelines require continuous ECG monitoring for deep sedation and general anesthesia, but its use for moderate sedation is suggested only for patients with significant cardiovascular disease. This would include patients with known rhythm disturbances, including those managed with implanted pacemakers.
About the Authors
Dr. Casabianca is a Assistant Professor in the Department of Anesthesiology and Surgery (Dentistry), University of Toledo College of Medicine, in Toledo, Ohio.
Dr. Becker is a Professor of Allied Health Sciences at Sinclair Community College and Associate Director of Education, General Dental Practice Residency, at Miami Valley Hospital in Dayton, Ohio.
REFERENCES
1. American Dental Association. ADA guidelines for the use of sedation and general anesthesia by dentists. Available at: http://www.ada.org/prof/resources/positions/statements/anesthesia_guidelines.pdf. Accessed March 2, 2009.
2. American Society of Anesthesiologists Task Force on Sedation and Analgesia by Non-Anesthesiologists. Practice guidelines for sedation and analgesia by non-anesthesiologists. Anesthesiology. 2002;96:1004-1017.
3. Becker DE, Casabianca AB. Respiratory monitoring: technical and physiological considerations. Anesth Prog. 2009;56:14-22.
4. Morgan GE Jr, Mikhail MS, Murray MJ. Patient monitors. In: Clinical Anesthesiology. 4th ed. New York: McGraw-Hill; 2006.
5. Ganong WF. Dynamics of blood & lymph flow. In: Review of Medical Physiology. 22nd ed. McGraw-Hill; 2005.
6. Guyton AC, Hall JE. Textbook of Medical Physiology. 11th ed., Philadelphia: Elsevier, 2006.
7. Norris EJ. Anesthesia for vascular surgery. In: Miller RD, ed. (2005). Miller’s Anesthesia. 6th ed., Philadelphia: Churchill Livingstone; 2005.
8. Becker DE. Fundamentals of ECG interpretation. Anesth Prog. 2006;53:53-64. Anesth Prog. 56:53-61.