You must be signed in to read the rest of this article.
Registration on CDEWorld is free. Sign up today!
Forgot your password? Click Here!
Pain is a complex experience consisting of a specific sensation and the reactions evoked by that sensation. Conventional analgesics either interrupt ascending nociceptive impulses or depress their interpretation within the central nervous system (CNS). A variety of so-called ‘‘analgesic adjuncts’’ have proven efficacy for managing chronic pain but will not be addressed in this article. They include various antidepressants and anticonvulsants that either enhance descending inhibitory pathways or modulate excitatory neural traffic that amplifies pain interpretation. These agents have marginal benefit in the management of acute pain, and they are not regarded as ‘‘analgesics’’ in the conventional sense.
Analgesics are classified as opioids and nonopioids, but dated terms like narcotic and non-narcotic are used interchangeably. Formerly, it was believed that opioids acted only within the brain and spinal cord, but the action of nonopioids was confined to the periphery (ie, the site of injury). This explanation is no longer tenable, however; both are known to act centrally and peripherally.1,2 In fact, the feature that best distinguishes these analgesic classes is their mechanism of action. Opioids activate specific receptors in a manner identical to opiates, such as morphine. Nonopioids interrupt prostaglandin synthesis, thereby resembling aspirin in action.
NONOPIOID ANALGESICS
The nonopioid analgesics include acetaminophen (APAP) and the nonsteroidal anti-inflammatory drugs (NSAIDs). The analgesic efficacy of these agents is typically underestimated. This is unfortunate because they generally are equivalent or superior to opioids for managing musculoskeletal pain, and they produce a lower incidence of side effects, including the potential for abuse. Dental pain is included in the musculoskeletal category, and for decades studies have repeatedly found that NSAIDs are generally superior to opioids at conventional dosages.3-5 This principle will be revisited during the final portion of this article, but at this time it is important to review essential pharmacological features of the nonopioids.
NSAIDS
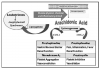
Actions and Effects. Ibuprofen is conventionally regarded as the prototype of this large group of synthetic compounds known for their analgesic, antipyretic, and anti-inflammatory efficacy. These therapeutic effects and their most notable side effects can be explained almost entirely by their ability to inhibit the cyclooxygenase (COX) required for synthesis of various families of prostanoids.6 This action is illustrated and further explained in Figure 1.
Precautions and Side Effects. Clinical use of NSAIDs is predicated on their ability to reduce the synthesis of prostaglandins implicated in pain, fever, and inflammation. However, these agents are hardly selective in this goal and also inhibit the production of additional prostanoids that perform useful physiological functions. This accounts for potential side effects and contraindications.
The most frequent side effects of NSAIDs are related to their gastrointestinal (GI ) toxicity. Prostaglandins stimulate the production of a mucous lining that protects the stomach and small intestine. The erosive and ulcerative side effects common to NSAIDs are attributed to their inhibiting the synthesis of these particular prostaglandins. This action not only occurs locally as orally administered drug lies in contact with gastric mucosa but also follows absorption and systemic distribution to the GI mucosa. Parenteral administration does not preclude a risk for GI erosions and ulcerations. It is important to distinguish dyspepsia (upset stomach) from GI toxicity, which reflects actual mucosal damage. The incidence of dyspepsia attributed to NSAIDs does not correlate with mucosal injury. Although less likely to produce gastric upset, buffered aspirin carries similar risk for mucosal damage as regular aspirin.7
The ability of NSAIDs to inhibit cyclooxygenases in platelets reduces the synthesis of thromboxane A2, which normally contributes to platelet aggregation. This accounts for the so-called antiplatelet effect of these agents and is a consideration following surgical procedures. However, aspirin is the only NSAID that has proven effective in preventing thrombotic events such as acute coronary syndromes or stroke. This is so because the antiplatelet action of aspirin is irreversible, lasting the life span of the platelet (10 to 14 days). Other NSAIDs bind weakly and reversibly to platelet cyclooxygenases, which results in loss of their mild antiplatelet influence after drug elimination.8 Although non-aspirin NSAIDs all prolong bleeding times to some degree, this does not correlate with significant clinical bleeding following minor surgical procedures. However, non-aspirin NSAIDs generally are withheld before major thoracic, abdominal, or orthopedic procedures. If aspirin is medically necessary for patients, such as those with endovascular stents who are at risk for life-threatening clot formation, it should not be withdrawn.
NSAIDs should be avoided in patients who suffer bleeding disorders and in those taking anticoagulants such as warfarin and powerful antiplatelet drugs such as clopidogrel (Plavix). Patients receiving monotherapy with low-dose aspirin are not as great a concern but should be considered. The issue with NSAIDs is due not so much to their antiplatelet action but to NSAID-induced injury of GI mucosa that may bleed far more profusely in this patient population. Aspirin provides a maximum antiplatelet influence at a very low dose—80 mg daily—and frequently is prescribed in combination with warfarin without consequence because such doses have a lower potential to produce gastric insult. In contrast, other NSAIDs increase the risk for GI bleeding twofold to threefold in patients medicated with clopidogrel (Plavix) and fourfold to fivefold in those taking warfarin.9 All concerns related to NSAID-induced mucosal injury are particularly important for older patients, especially those taking antithrombotic medications, including low-dose aspirin.
Prostaglandins play an essential role in renal perfusion, and diminished levels of these are believed to account for reported cases of nephrotoxicity after long-term NSAID use. In the healthy patient, nephrotoxicity attributed to NSAIDs requires high doses for extended periods (eg, a year or longer).10 However, a patient with compromised renal function relies more heavily on prostaglandins for adequate function, and acute renal failure can occur within 24 hours of NSAID administration. NSAIDs must never be prescribed for patients who have known or questionable renal function. The ability of NSAIDs to alter renal function has resulted in concern regarding their long-term use in patients with hypertension and heart failure.11,12 Altered renal function accompanies the pathogenesis of these disorders, and any further renal decline may exacerbate their condition. This concern has not been found relevant with short-term NSAID use (eg, 5 to 7 days).
By inhibiting cyclooxygenase, NSAIDs shunt the arachidonic pathway toward leukotriene synthesis (Figure 1). Leukotrienes mediate a variety of tissue responses, including those associated with bronchospasm and anaphylaxis.13 Certain individuals may be extremely sensitive to even subtle elevation in leukotriene synthesis, which may result in signs and symptoms of allergic response. It is recommended that the term aspirin or NSAID intolerance should be used to distinguish this reaction from true hypersensitivity responses mediated by immunoglobulin (Ig)E. Acetaminophen is the conventional alternative for patients reporting an allergic reaction to any NSAID, unless the patient can identify a particular product that he or she has tolerated without problem in the past.
In summary, NSAIDs are contraindicated for patients who have a current history of nephropathy, erosive or ulcerative conditions of the GI mucosa, anticoagulant therapy, hemorrhagic disorders, or intolerance or allergy to any NSAID. They also should be avoided during pregnancy because prostaglandins maintain patency of the ductus arteriosus during fetal development. Although this concern is most relevant during the third trimester, NSAIDs generally should be avoided throughout pregnancy. In all cases where NSAIDs are contraindicated, acetaminophen is the conventional nonopioid alternative.
Drug Interactions. After prolonged use, NSAIDs may interfere with the effectiveness of most classes of antihypertensive medications; calcium channel blockers are a notable exception. The precise mechanism for this interaction is unknown but is believed to be related to diminished vasodilator actions attributed to renal prostaglandins. In the rare event that postoperative analgesics must be continued for longer than 5 days, hypertensive patients should return to the office for blood pressure assessment. If pressure has elevated more than 10% above baseline, it would be wise to replace the NSAID with acetaminophen.
Ibuprofen has been found to competitively inhibit the antiplatelet influence of aspirin.14,15 It is the only NSAID implicated in this interaction, but diclofenac and the selective COX-2 inhibitors are the only agents that have been confirmed not to interact.15 An empiric solution to this problem is predicated on the fact that the antiplatelet influence of low-dose aspirin occurs when it contacts platelets within the hepatic portal system after absorption.16 Simply advise the patient to take daily aspirin upon arising and to delay the first dose of ibuprofen for 1 to 2 hours. By this time, the antiplatelet influence of aspirin will have been established.17 This entire issue may eventually prove moot because its actual clinical relevance has been challenged impressively. Cryer et al18 found that thromboxane inhibition by aspirin was reduced by only 1% after 10 days of concurrent ibuprofen use, and Patel19 found no increase in incidence of myocardial infarction over a 10-year period in patients with coronary disease taking ibuprofen with low-dose aspirin.
Recently concern has been introduced regarding increased risk for GI mucosal injury in patients taking selective serotonin reuptake inhibitor (SSRI) antidepressants and NSAIDs. This risk is most significant after prolonged use of NSAIDs, but caution may be advised during short-term use for patients who have a previous history of mucosal injury.20,21 Finally, serum levels of lithium and methotrexate are elevated during concurrent consumption of NSAIDs. To prevent toxicity, NSAIDs should be avoided in patients medicated with these agents, particularly those taking high-dose regimens.
Therapeutic Uses. In general, no convincing evidence indicates that a particular NSAID is more effective or safer than other members of this drug class.22 Selective COX-2 inhibitors such as celecoxib produce less GI toxicity after short-term use, but this advantage wanes as consumption continues. Chou et al23 published an impressive evidence-based analysis of NSAIDs for the Oregon Evidence-Based Practice Center that supports this generalization. Nevertheless, patients vary considerably in their clinical response and GI tolerance to a particular agent. Given its unsurpassed efficacy and low side effect profile and cost, ibuprofen is generally a sound initial choice. Regardless of the agent selected, however, an optimal dosing schedule should be maintained for 2 to 3 days before an alternative agent is prescribed. It is reasonable to select an alternative NSAID for initial therapy for patients who appear to question the effectiveness of a product that is available over-the-counter. Regardless of the NSAID selected, clinical considerations are identical.
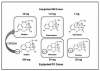
All NSAIDs have greater potency as analgesics and antipyretics than as anti-inflammatory agents; higher doses are required to achieve anti-inflammatory than analgesic effects. This may reflect a different site of action for analgesic versus anti-inflammatory actions (eg, CNS vs periphery), but this has not been confirmed. For example, a single 200-mg to 400-mg dose of ibuprofen may reduce pain and fever, but daily consumption of 1600 mg to 2400 mg may be required to suppress inflammation adequately. Nevertheless, if we consider only their analgesic properties, the dose-response curves for nonopioids (NSAIDs and acetaminophen) exhibit a ceiling effect; additional increases in dose provide no further benefit (Figure 2). The ceiling responses for aspirin and acetaminophen occur at 1000 mg, and the analgesic ceiling for ibuprofen is achieved at 400 mg.24,25 As the dose of an NSAID is increased, anti-inflammatory effects improve until maximum safe doses preclude any further increase. Most NSAIDs have ranges in their recommended dosages, but precise doses for their analgesic ceilings have not been determined. It is reasonable to presume that the lower doses, like those for ibuprofen, confer analgesia, and the higher range adds anti-inflammatory efficacy. With this in mind, lower dosages should be selected for noninflammatory pain, and higher ranges are reserved for those situations in which inflammation and swelling are significant cofactors. Of course most cases of dental pain have at least some degree of contributing inflammation.
Preoperative use of NSAIDs has been demonstrated repeatedly to decrease the intensity of postoperative pain and swelling.26,27 This is not surprising because NSAIDs inhibit the ‘‘formation’’ of prostaglandins; however, they do not destroy or inhibit those already formed. More recent understanding of pain mechanisms reveals that benefits of this practice are evident so long as prostaglandin synthesis is inhibited before local anesthesia wanes. Otherwise, prostaglandins trigger nociceptive impulses that travel to the brain and ‘‘wind up’’ the brain’s interpretation of pain intensity. When an extensive surgical procedure is planned, optimal serum levels of an NSAID should be established preoperatively or before patient discharge, while tissues remain anesthetized. This ‘‘preemptive analgesia’’ may be useful for endodontic and extensive restorative procedures as well.
COX-2 Inhibitors
As was stated previously, clinical trials comparing the COX-2 inhibitors (eg, celecoxib [Celebrex]) versus conventional NSAIDs have not identified substantive differences in their anti-inflammatory or analgesic efficacy.22,23 Clinical studies have found celecoxib less effective as an analgesic when compared with ibuprofen and naproxen.22 COX-2 agents offer the advantages of no increase in bleeding time and minimal GI injury despite a comparable incidence of dyspepsia. However, controversy persists regarding their risk for thrombotic events in patients with atherosclerotic disease. As is illustrated in Figure 1, selective COX-2 inhibition tilts prostanoid production toward platelet aggregation. Indeed several publications have suggested an increase in acute coronary events in patients with preexisting CAD who have been medicated with COX-2 inhibitors. In fairness, this correlation has been found with most of the nonselective NSAIDs as well. Naproxen is the only NSAID that appears to lack this correlation. Nevertheless, it is probably wise to avoid selective COX-2 inhibitors in patients with significant atherosclerotic disease.
Acetaminophen
Actions and Effects. Compared with NSAIDs, the mechanism of action of acetaminophen is less clear but is believed to involve an inhibition of prostaglandin synthesis within the CNS.28 It has little influence on peripheral prostaglandin synthesis, especially within inflamed tissues.7 This is a likely explanation for its lacking anti-inflammatory efficacy and sharing none of the peripheral side effects attributed to NSAIDs. However, it is an ideal analgesic for patients who present any contraindications to NSAIDs. As an analgesic and antipyretic, acetaminophen is equal in potency and efficacy to aspirin29 and presumably may be somewhat inferior to ibuprofen and other NSAIDs as well. Hepatotoxicity is the most significant adverse effect of acetaminophen. It is attributed to a toxic metabolite that cannot be adequately conjugated when dosages exceed 200 mg to 250 mg/kg in a 24-hour period.30 The dose may be less for patients who are poorly nourished, who have liver dysfunction, or who are being treated with other hepatotoxic medications. For example, in contrast to the 4 g/d allowed healthy patients, those suspected of chronic alcoholism should limit their maximum daily intake to 2 grams.
Summary of Nonopioids
Most cases of postoperative dental pain include an inflammatory component. For this reason, NSAIDs are the most rational first-line agents—often superior to conventional dosages of opioids. Should a patient present a contraindication to NSAIDs, acetaminophen is the only alternative. Nonopioids exhibit a ceiling to their analgesic response, but optimal doses should be established before it is assumed that the NSAID has failed. Furthermore, the combination of an NSAID with acetaminophen provides greater analgesic efficacy than does either agent alone,31 and this strategy may obviate the need for opioids. Data relevant for prescribing the most commonly used nonopioids are summarized in Table 1.
OPIOID ANALGESICS
Actions and Effects
Opioids produce most of their therapeutic and adverse effects by acting as agonists at opioid receptors. Scientists have not entirely established the physiological significance of these receptors, but they are activated by a variety of endogenous ligands, collectively called endorphins. Opioid receptors germane to clinical practice are located within the CNS, but peripheral receptors have also been characterized.2 Unlike nonopioids, which exhibit a ceiling analgesic response, opioids demonstrate greater efficacy as the dose is increased (Figure 2). Unfortunately, when pain is very severe, side effects may preclude the use of doses adequate to produce complete analgesia.
Only two of the five opioid receptors isolated thus far have relevance for clinical practice. The effects mediated by mu and kappa receptors are summarized in Table 2. Morphine produces its effects by acting as an agonist at both mu and kappa receptors, while naloxone acts as an antagonist.32
The mu receptor is responsible for mediating analgesia and two of the most undesirable side effects attributed to opioids: respiratory depression and dependence. Mu effects have unlimited intensity, increasing proportionately with dose. Therefore, a striking contrast exists between the unlimited analgesic efficacy of mu agonists and the limited or ceiling effect described for the nonopioids.
Like mu receptors, the kappa receptor mediates analgesia and respiratory depression, but efficacy at this receptor is limited.28 These two receptors provide comparable efficacy after doses equivalent to 10 mg morphine IM, but the response from kappa receptors does not increase with greater doses. When high doses of opioids are used, selective kappa agonists are viewed as safer, but less analgesic, compared with traditional mu agonists.
Knowledge regarding the kappa receptor spawned the synthesis of several novel compounds that act as agonists at kappa receptors but act as antagonists at mu receptors. Nalbuphine (Nubain) is an example (Table 2). These so-called agonist-antagonists are not constipating, produce less respiratory depression at higher doses, and have less potential for abuse, but their limited analgesic efficacy diminishes their value when postoperative pain is severe. Higher doses are no more effective than conventional doses. Because they act as antagonists at mu receptors, agonist-antagonists may precipitate a withdrawal syndrome in patients dependent on opioids. They are good choices for patients who have a previous history of drug seeking, but they must never be given to a patient who is currently dependent. Dysphoric reactions produced by these agents were formerly believed to be mediated by sigma receptors. However, this receptor is no longer considered an opioid receptor, and dysphoria is credited as a kappa receptor phenomenon.32
Dependence, Tolerance, and Addiction
Fear of dependence and addiction often results in underprescribing of opioids for severe acute, chronic, and even terminal pain. This unfortunate practice is due to poor understanding of dependence, tolerance, and addiction.
Dependence occurs when the body accommodates to the influences of a drug and, upon sudden discontinuation, the patient experiences a withdrawal syndrome that generally includes reactions opposite those produced by the particular drug. For example, opioids produce sedation, lethargy, and constipation. A patient who is experiencing opioid withdrawal becomes excited and experiences abdominal cramping and diarrhea. If opioid doses are tapered gradually, a dependent patient will not experience withdrawal. Patients who consume opioids regularly for longer than a week can develop some degree of dependence. This may require gradual tapering of the dosage to avoid withdrawal symptoms, which can be confused as an exacerbation of the painful condition. However, this does not mean that the patient has become addicted.22,32
After repeated administration, patients develop tolerance to opioids. This is to say that greater doses are required to produce the same intensity of effect formerly provided by a smaller dose. Tolerance to analgesia, sedation, and respiratory depression occurs simultaneously, but it is curious that no tolerance occurs to the constipating or miotic effects of opioids. This is problematic for the patient with chronic or terminal pain. Although staggering doses may be required to control pain and generally will not jeopardize the patient’s respiratory status, constipation can become extremely severe, and night vision becomes impaired. Similar doses, if administered to patients who have not developed tolerance (ie, opioid-naive patients), would certainly be lethal. These identical issues must be considered when one is managing dental pain for patients who are chronic opioid abusers.
Addiction is distinct from dependence or tolerance. It is a compulsive behavior centered on seeking a drug and its effects for nonmedical reasons—generally for pleasure. It is a complex psychiatric phenomenon, but it should not be attributed to the drug. Addictive behavior can be reinforced by a particular drug, but it is not a pharmacodynamic property. A patient who lacks addictive behavior can be easily weaned from opioid dosages without fear of precipitating addictive behavior. In contrast, an addicted patient will seek the drug despite having no remaining evidence of dependence or medical need for the drug. Opioids produce dependence, even after as little as 5 to 7 days of therapy, and this may require institution of a tapering dosage schedule. However, opioids do not produce addiction; they should not be withheld on the presumption that the patient will become ‘‘addicted.’’32 Obviously, opioids must be prescribed cautiously for patients who demonstrate addictive personality.
Therapeutic Considerations
Despite recent scientific literature to the contrary, many believe that certain opioids are more effective or more dangerous than others. This is simply not the case; equipotent doses are equianalgesic. When administered subcutaneously or intramuscularly, 10 mg morphine, 75 mg meperidine, and 120 mg codeine all produce equivalent analgesia and side effects.32 However, as will be explained below, issues regarding metabolism and activity of metabolites have been noted with some of these agents.
After oral administration, gastric degradation and first-pass metabolism require that larger doses be used if one is to achieve analgesia comparable with that following parenteral administration. For example, the oral-to-parenteral dose ratio for morphine is generally regarded as 3:1. If one is to duplicate the analgesic efficacy of a standard 10-mg IM injection of morphine, a 30-mg oral dose must be prescribed. Equianalgesic doses of commonly used opioids are found in Table 3.
Effective pain control is predicated on selecting an optimal dose, rather than selecting a particular agent. However, individual differences in patient response and pharmacokinetic differences (eg, duration, elimination half-life) may favor the use of a particular agent. Morphine 7.5 mg to 10 mg IM is a relatively safe and common standard for inpatient analgesia. By using this as a reference point, one can select equianalgesic doses of other agents for both parenteral and oral regimens. The opioid doses listed in Table 3 are equianalgesic and present equivalent risks for serious side effects.
Considerations for Specific Opioids
Codeine has very little affinity for the mu receptor and may be considered a prodrug because 10% of the parent drug is converted to morphine by cytochrome P450 CYP2D6.The morphine metabolite accounts for its entire analgesic effect32 (Figure 3). Altered activity of CYP2D6 offers one explanation for varied responses to codeine and to its derivatives that will be addressed below. Roughly 5% to 10% of the Caucasian population metabolizes codeine poorly because these individuals have inherited two nonfunctional alleles for synthesis of CYP2D6. For them, analgesia resulting from codeine will be less than expected with the general population. It has also been estimated that 1% to 7% of Caucasians have elevated CYP2D6 activity, and this may account for heightened sensitivity.33 Likewise, a variety of drugs that a patient may be taking concurrently have the ability to inhibit or induce CYP2D6 activity. For example, the SSRI antidepressants are CYP2D6 inhibitors, making codeine less effective. This is established for fluoxetine (Prozac) and paroxetine (Paxil) but appears less likely with other agents of this class.34 In contrast, dexamethasone is a CYP2D6 inducer and will enhance the portion of codeine demethylated to morphine.
Hydrocodone and Oxycodone. Hydrocodone and oxycodone are more attractive analgesics than codeine. They also are methylated, but these parent drugs appear to have better affinity for opioid receptors than codeine. Hydrocodone is demethylated to hydromorphone in quantities sufficient to credit both the parent drug and this active metabolite with its analgesic influence. For this reason, hydrocodone shares the same considerations regarding demethylation addressed previously for codeine.33 In contrast, the analgesic effect of oxycodone is almost entirely attributed to the parent drug because only scant amounts are demethylated to oxymorphone.33 This makes it the better choice for patients taking medications known to inhibit CYP2D6.Their potency allows for lower doses of these agents and reduces the incidence of nausea compared with codeine.
Unfortunately, equianalgesic doses for these codeine derivatives were poorly understood initially, which spawned release of combination products that contain irrational dosage formulations. Equipotent doses listed in textbooks vary somewhat, but those provided in Table 3 are well accepted and indicate that 200 mg codeine, 30 mg hydrocodone, and 20 mg oxycodone are equipotent oral doses, and these are equianalgesic to the conventional opioid standard of morphine 10 mg IM or 30 mg PO. Codeine doses have been well studied, and from this table we find that the oral dose for codeine is approximately 20 times the IM dose of morphine (200 mg vs 10 mg). Clinical studies that have attempted to address equianalgesic doses of codeine derivatives are sparse, but they support this same ratio. Beaver et al35 found that oxycodone 10 mg was comparable with codeine 100 mg, and this would extrapolate to oxycodone 20 mg and codeine 200 mg. Studies by Hopkinson36 and by Beaver37 have shown that hydrocodone 10 mg was approximately equipotent to codeine 60 mg, and this would extrapolate to 33 mg hydrocodone and 200 mg codeine. It is not uncommon for patients to report previous episodes of nausea as an ‘‘allergic reaction.’’ However, IgE antibodies have been detected that react with several opioids, including codeine,38 and nearly all opioids are capable of triggering degranulation of mast cells leading to the direct release of histamine.39 Until issues regarding cross-reactivity among opioids are resolved, a prudent approach would be to select alternatives that are molecularly dissimilar. For example, when a patient reports clinical signs that are allergic in nature, one should select an agent that is not derived from morphine or codeine (eg, propoxyphene, pentazocine).40
Meperidine. Meperidine 75 mg to 100 mg is equianalgesic to morphine 10 mg after IM administration. A significant portion of an IM dose of meperidine is converted to normeperidine, a metabolite that has no analgesic properties but is a noted cardiovascular and CNS stimulant. Furthermore, this metabolite has a 15- to 20-hour elimination half-life, compared with 3 hours for the parent drug.32 For hospitalized patients, meperidine is used for only a day or two; otherwise, normeperidine will accumulate. In fact, many hospitals have deleted it from their formularies. This issue becomes even more problematic after oral administration in outpatients. Oral bioavailability for meperidine is approximately 25%, which requires a 300-mg dose to be equianalgesic to its IM dose of 75 mg. This introduces an even greater risk for accumulation of normeperidine. Poor oral absorption and accumulation of normeperidine make meperidine a very poor choice as an oral analgesic.
Propoxyphene. Propoxyphene is available only for oral administration. The equianalgesic dose compared with morphine has not been established, but its potency is low. By convention, 100 mg is considered equipotent to oral codeine 60 mg. It is similar to meperidine in that it is converted to norpropoxyphene, a stimulant that has an elimination half-life of 30 hours.32 Its use should be limited to short-term management of mild to moderate pain.
Pentazocine. Pentazocine is the only oral agonist-antagonist analgesic available in the United States. It produces its analgesic effect by acting as an agonist at kappa receptors but is an antagonist at mu receptors. Therefore it reverses all effects of traditional mu agonist opioids if taken concurrently. Unlike mu agonists, which provide unlimited analgesic efficacy, kappa agonists exhibit a ceiling to their analgesic effect, and no benefit is derived by increasing doses beyond 50 mg. Pentazocine is available in the United States for oral use compounded with naloxone to prevent parenteral injection abuse issues. If injected, naloxone will block all effects of pentazocine, rendering it useless. When taken by mouth, however, naloxone has no oral bioavailability and will not hinder pentazocine actions. Additionally, pentazocine is available compounded with APAP. It should not be used in the presence of other opioids. When other opioids are present, pentazocine will serve as an opioid antagonist, thus reducing the patient’s analgesia. Additionally, it should not be prescribed for patients who are opioid dependent and at risk for withdrawal. It is an attractive choice for patients who have a previous history of opioid abuse because it does not provide euphoric effects mediated by conventional mu agonists. Because it is not a mu receptor agonist, constipation is unlikely.
Tramadol. Tramadol is a centrally acting analgesic with binary action. It is not classified as a controlled substance in the United States. The parent drug inhibits the reuptake of norepinephrine and serotonin. This resembles the action of tricyclic antidepressants and potentiates descending neural pathways that inhibit incoming nociceptive impulses. This action has proven efficacy in the management of chronic pain. However, any benefit for tramadol in acute postoperative pain management is not as well defined. The principal metabolite of tramadol, O-desmethyltramadol (M1), demonstrates agonist action on mu receptors, providing analgesic efficacy approximating that of codeine 60 mg. Formation of this metabolite is provided by CYP2D6 enzymes and introduces the identical risk for drug interactions described earlier for codeine. Tramadol is not recommended for patients with a tendency toward opioid abuse or dependence.41 It is available in combination with acetaminophen but is no more effective than codeine-acetaminophen combinations.42
SELECTING ANALGESIC REGIMENS
Mild to moderate pain generally can be managed by using optimal doses of nonopioids: ibuprofen 400 mg to 800 mg, acetaminophen 1000 mg, or a combination of the two. Although it is unwise to combine NSAIDs, the addition of acetaminophen to an NSAID is reasonable because they have different sites for their analgesic action.31,43,44 Regardless of pain severity, one should seek to optimize ‘‘around-the-clock’’ dosages of these agents and then, if necessary, add an opioid to the regimen as needed for breakthrough pain. This practice generally will reduce the amount of opioid required, sometimes to only a fraction of the maximum doses listed in Table 3. It is irrational to prescribe opioid combinations routinely as ‘‘first-line’’ analgesics.
To further focus on the importance of nonopioid analgesics, refer to the graph provided as Figure 4. These data were derived from a clinical study funded by a drug company to promote a combination product containing ibuprofen and oxycodone.5 It is significant that the analgesia provided by single-entity agents reconfirms clinical studies published repeatedly over the past 3 decades. NSAIDs are more effective for musculoskeletal pain than are conventional doses of opioids. In fact, oxycodone 5 mg (the dose combined with APAP in the most commonly prescribed Percocet tablet) is no more effective than placebo. Notice, however, that a small increment of opioid (oxycodone 5 mg) added to ibuprofen improves pain relief. This illustrates the fact that opioids are synergistic, and analgesia can be improved by titrating additional opioid increments to an optimally dosed nonopioid.
It is not surprising that such a large number of commercially compounded analgesics containing both nonopioid and opioid ingredients have been produced. The opioid contained in most of these products is either hydrocodone or oxycodone. Some of these combinations appear to have been formulated with little consideration given to equianalgesic dosage strategies. Additionally, several products contain large quantities of acetaminophen that preclude the use of multiple tablets to achieve an adequate amount of opioid for patients who experience severe pain. When prescribing combination products, the clinician must pay particular attention to the amount of acetaminophen used separately or compounded so that the maximum daily dose of 4 grams is not exceeded. In many cases, it is better to write separate prescriptions for the opioid and the nonopioid at dosages that more precisely address the analgesic needs of the patient. Suggested regimens are presented in Table 4.
The dentist must be cautious when prescribing for patients managed over the long term with opioids by their physician. Ideally, these patients have contracted with their physician to decline opioid prescriptions from other healthcare providers.45 Regardless of their arrangements, the dentist should avoid increasing the current opioid dose or prescribing additional opioids for postoperative pain control. The patient’s daily nonopioid regimen should be optimized, and, if opioids are required, the physician should be asked to temporarily increase the dosage. Pain experienced after dental surgery is additive to the patient’s normal chronic intensity of pain, and opioid tolerance may require a temporary increase in opioid dosage.
SUMMARY
Careful selection of an effective analgesic regimen should be based on the type and amount of pain the patient is expected to experience. This strategy can prevent the stress and anxiety associated with breakthrough pain.46-48 When analgesics fail, it is not unusual for patients to make desperate attempts to seek relief. The clinician should develop several safe and effective analgesic regimens based on estimates of anticipated pain intensity. Following are key features for the proper management of acute postoperative pain:
1. Patients benefit from receiving optimal NSAID doses given at regular, ‘‘clock-based’’ time intervals. These agents are effective and relatively safe and reduce the need for opioids. In situations where pain can be anticipated, the analgesia may be optimized by commencing administration before local anesthesia wanes (ie, ‘‘preemptive analgesia’’).
2. Although NSAIDs achieve an analgesic ceiling at their lower dose ranges, it is proper to prescribe higher doses for most cases of dental pain to derive benefit from their anti-inflammatory properties.
3. The site of action of acetaminophen differs from that of NSAIDs. Therefore, the analgesic effect of acetaminophen is considered synergistic when combined with NSAIDs.
4. If the dose of an NSAID, acetaminophen, or their combination has been optimized but pain persists, an opioid should be added. A commercially available combination product containing opioid and acetaminophen may be an option and is easy to prescribe. However, when prescribing these combination products, the practitioner must be cautious not to exceed 4 grams of acetaminophen per day because of concerns about hepatic injury.
5. Because opioids have no ceiling dose, opioid dosing is better accomplished by prescribing it separately in some situations. This allows opioid to be titrated to the analgesic dose required and decreases concern for acetaminophen toxicity.
6. Avoid prescribing any opioid product for patients already receiving opioids for chronic pain disorders and for those under treatment for opioid abuse. It is appropriate to request an increase in dosage from the prescribing physician if necessary.
REFERENCES
1. Malmberg AB, Yaksh TL. Hyperalgesia mediated by spinal glutamate or substance P receptor blocked by spinal cyclooxygenase inhibition. Science. 1992;257:1276-1279.
2. Stein CS. The control of pain in peripheral tissue by opioids. N Engl J Med. 1995;332:1685-1690.
3. Forbes JA, Kehm CJ, Grodin CD, Beaver WT. Evaluation of ketorolac, ibuprofen, acetaminophen, and an acetaminophen-codeine combination in postoperative oral surgery pain. Pharmacotherapy. 1990;10:94S-105S.
4. Fricke JR Jr, Angelocci D, Fox K , et al. Comparison of the efficacy and safety of ketorolac and meperidine in the relief of dental pain. J Clin Pharmacol. 1992;32:376-384.
5. Van Dyke T, Litkowski LJ, Kiersch TA, et al. Combination oxycodone 5 mg/ibuprofen 400 mg for the treatment of postoperative pain: a double-blind, placebo- and active-controlled parallel-group study. Clin Ther. 2004;26:2003-2014.
6. Burke A, Smyth EM, FitzGerald GA. Analgesic-antipyretic agents: pharmacotherapy of gout. In: Brunton LL, Lazo JS, Parker KL, eds. Goodman and Gilman’s The Pharmacological Basis of Therapeutics. 11th ed. New York: McGraw-Hill; 2006.
7. Kimmey MB. Cardioprotective effects and gastrointestinal risks of aspirin: maintaining the delicate balance. Am J Med. 2004;117:72s-78s.
8. Goldenberg NA, Jacobson MT, Manco-Johnson MJ. Duration of platelet dysfunction after a 7-day course of ibuprofen. Ann Intern Med. 2005;142:506-509.
9. Delaney JA, Opatrny L, Brophy JM, Suissa S. Drug-drug interactions between antithrombotic medications and the risk of gastrointestinal bleeding. CMAJ. 2007;177:347-351.
10. DeBroe ME, Elseviers MM. Analgesic nephropathy. N Engl J Med. 1998;338:446-452.
11. Gislason GH, Rasmussen JN, Abildstrom SZ, et al. Increased mortality and cardiovascular morbidity associated with use of nonsteroidal anti-inflammatory drugs in chronic heart failure. Arch Intern Med. 2009;169:141-149.
12. White WB. Defining the problem of treating the patient with hypertension and arthritis pain. Am J Med. 2009;122(5 suppl):S3-S9.
13. Babu KS, Salvi SS. Aspirin and asthma. Chest. 2000;118:1470-1476.
14. Catella-Lawson F, Reilly MP, Kapoor SC, et al. Cyclooxygenase inhibitors and the antiplatelet effects of aspirin. N Engl J Med. 2001;345:1809-1817.
15. MacDonald TM, Wei L. Effect of ibuprofen on cardioprotective effect of aspirin. Lancet. 2003;361:573-574.
16. Patrono C. Aspirin as an antiplatelet drug. N Engl J Med. 1994;330:1287-1294.
17. Abramowicz M. Do NSAIDS interfere with the cardioprotective effects of aspirin? The Medical Letter on Drugs and Therapeutics. 2004;46:61-62.
18. Cryer B, Berlin RG, Cooper SA, Hsu C, Wason S. Double-blind, randomized, parallel, placebo-controlled study of ibuprofen effects on thromboxane B2 concentrations in aspirin-treated healthy adult volunteers. Clin Ther. 2005;27:185-191.
19. Patel TN, Goldberg KC. Use of aspirin and ibuprofen compared with aspirin alone and the risk of myocardial infarction. Arch Intern Med. 2004;164:852-856.
20. Yuan Y, Tsoi K, Hunt RH. Selective serotonin reuptake inhibitors and risk of upper GI bleeding: confusion or confounding? Am J Med. 2006;119:719-727.
21. Pinto A, Farrar JT, Hersh EV. Prescribing NSAIDs to patients on SSRIs: possible adverse drug interaction of importance to dental practitioners. Compend Contin Educ Dent. 2009;30:142-151; quiz 152, 154.
22. Abramowicz M, ed. Drugs for pain: treatment guidelines from The Medical Letter. The Medical Letter on Drugs and Therapeutics. 2007;5:23-32.
23. Chou R, Helfand M, Peteron K, et al. Drug Class Review on Cyclooxygenase (COX)-2 Inhibitors and Nonsteroidal Antiinflammatory Drugs (NSAIDs). Portland, Ore: Oregon Evidence-Based Practice Center, Oregon Health & Science University; 2006.
24. Troullos ES, Freeman RD, Dionne RA. The scientific basis for analgesic use in dentistry. Anesth Prog. 1986;33:123-138.
25. Laska EM, Sunshine A, Marrero I, et al. The correlation between blood levels of ibuprofen and clinical analgesic response. Clin Pharmacol Ther. 1986;40:1-7.
26. Dionne RA, Campbell RA, Cooper SA, et al. Suppression of postoperative pain by preoperative administration of ibuprofen in comparison to placebo, acetaminophen, and acetaminophen plus codeine. J Clin Pharmacol. 1983;23:37-43.
27. Jackson DL, Moore PA, Hargreaves KM. Preoperative nonsteroidal anti-inflammatory medication for the prevention of postoperative dental pain. J Am Dent Assoc. 1989;119:641-647.
28. Piletta P, Porchet HC, Dayer P. Central analgesic effect of acetaminophen but not aspirin. Clin Pharmacol Ther. 1991;49:350-354.
29. Cooper SA. Comparative analgesic efficacies of aspirin and acetaminophen. Arch Intern Med. 1981;141:282-285.
30. Whitcomb DC, Block GD. Association of acetaminophen hepatotoxicity with fasting and ethanol. JAMA. 1994;272:1845-1850.
31. Hyllested M, Jones S, Pedersen JL, Kehlet H. Comparative effect of paracetamol, NSAIDs or their combination in postoperative pain management: a qualitative review. Br J Anaesth. 2002;88:199-214.
32. Gutstein HB, Akil H. Opioid analgesics. In: Brunton LL, Lazo JS, Parker KL, eds. Goodman and Gilman’s The Pharmacological Basis of Therapeutics. 11th ed. New York: McGraw-Hill; 2006.
33. Smith HS. Opioid metabolism. Mayo Clin Proc. 2009;84:613-624.
34. Abramowicz M, ed. Drug interactions. The Medical Letter. 1999;41:61-62.
35. Beaver WT, Wallenstein SL, Rogers A, Houde RW. Analgesic studies of codeine and oxycodone in patients with cancer. I. Comparisons of oral with intramuscular codeine and of oral and intramuscular oxycodone. J Pharmacol Exp Ther. 1978;207:92-100.
36. Hopkinson JH. Hydrocodone—a unique challenge for an established drug: comparison of repeated or doses of hydrocodone (10mg) and codeine (60mg) in the treatment of postpartum pain. Curr Ter Res. 1978;24:503-516.
37. Beaver WT, McMillan D. Methodological considerations in the evaluation of analgesic combinations: acetaminophen (paracetamol) and hydrocodone in postpartum pain. Br J Clin Pharmacol. 1980;10:215s-223s.
38. Harle DG, Baldo BA, Coroneos NJ, Fisher MM. Anaphylaxis following administration of papaveretum. Case report: implication of IgE antibodies that react with morphine and codeine, and identification of an allergenic determinant. Anesthesiology. 1980;71:489-494.
39. Weiss ME, Adkinson NF, Hirshman CA. Evaluation of allergic drug reactions in the perioperative period. Anesthesiology. 1989;71:483-486.
40. Becker DE. Management of allergic and pseudoallergic reactions. Dent Clin North Am. 1995;39:577-586.
41. Cicero TJ, Adams EH, Geller A, et al. A postmarketing surveillance program to monitor Ultram (tramadol) abuse in the United States. Drug Alcohol Depend. 1999;57:7-22.
42. Moore PA, Crout RJ, Jackson DL, et al. Tramadol hydrochloride: analgesic efficacy compared with codeine, aspirin with codeine, and placebo after dental extraction. J Clin Pharmacol. 2003;38:554-560.
43. Breivik EK, Barkvoll P, Skowlund E. Combining diclofenac with acetaminophen or acetaminophen-codeine after oral surgery: a randomized, double-blind, single-dose study. Clin Pharmacol Ther. 1999;66:625-635.
44. Issioui T, Klein KW, White PF, et al. The efficacy of premedication with celecoxib and acetaminophen in preventing pain after otolaryngologic surgery. Anesth Analg. 2002;94:1188-1193.
45. Chou R, Fanciullo GJ, Fine PG, et al. Opioid treatment guidelines: clinical guidelines for the use of chronic opioid therapy in chronic noncancer pain. J Pain. 2009;10:113-130.
46. Apfelbaum JL, Chen C, Mehta SS, Gan TJ. Postoperative pain experience: results from a national survey suggest postoperative pain continues to be undermanaged. Anesth Analg. 2003;97:534-540.
47. Mehlisch DR. The efficacy of combination analgesic therapy in relieving dental pain. J Am Dent Assoc. 2002;133:861-871.
48. Becker DE, Phero JC. Drug therapy in dental practice: nonopioid and opioid analgesics. Anesth Prog. 2005;52:140-149.