You must be signed in to read the rest of this article.
Registration on CDEWorld is free. Sign up today!
Forgot your password? Click Here!
Moderate and deep sedation is frequently required to manage the apprehensive dental patient. Clinicians and researchers alike continue the search for a “magic potion,” operating within the paradigm that an answer somehow lies in the selection of a perfect drug or drug combination. Suggestions have included a staggering number of single and multiple drug regimens, as well as alternative routes for their administration, including rectal and intranasal. None have achieved universal acceptance because no single drug or combination is suitable for all individuals and clinical situations. Most sedative agents can calm and sedate patients adequately, provided a sufficient concentration is achieved within the targeted neural tissues. An understanding of the challenge of achieving an effective but safe drug concentration in the brain rests on an appreciation of fundamental principles of pharmacokinetics.
Pharmacodynamic issues, drug actions and effects, will be the topic of a subsequent continuing education article. Pharmacokinetic processes include drug absorption, biotransformation (metabolism), distribution, and elimination. These processes are summarized in the Table. It is important that we distinguish each of these processes when administering medications by various routes.
Bioavailability
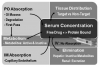
Bioavailability refers to the portion of an administered dose that reaches the systemic circulation in active form, and is thereby available for distribution to target tissues. Variables that influence drug bioavailability do not make this a simple matter, however. Consider the following questions while viewing Figure 1. What concentration of drug is required within the targeted tissue (biophase) for a given patient, on a given day, for a given level of anxiety? Furthermore, what serum concentration will provide and sustain this target concentration? Sedative serum concentrations for midazolam range from 30 ng/mL to 100 ng/mL.1 Provided we accept these data, what dose must we administer to achieve and sustain this precise concentration?
Bioavailability following intravenous (IV) administration is 100%; no absorption is required and the entire dose enters the systemic circulation. In contrast, the bioavailability of a drug administered orally (PO) is highly variable. A portion of the administered dose may not even be absorbed through the intestinal mucosa due to incomplete dissolution of the tablet, gastric degradation, and poor lipid solubility. Absorption requires some degree of lipid solubility if the drug is to diffuse through the epithelium of gastrointestinal mucosa. Furthermore, the portion actually absorbed enters the hepatic portal system and circulates to the liver where it may be subjected to first-pass metabolism before entering the systemic circulation. A drug administered by intramuscular (IM) injection is not subjected to degradation or first-pass influences, and the loosely joined capillary endothelium within a muscle allows drug diffusion regardless of lipid solubility. Nevertheless, the rate of absorption in muscle will vary and precludes any assurance that a prescribed dose will achieve an adequate peak serum concentration. All of these issues are obviated when sedatives are titrated intravenously.
Consider midazolam (Versed) as an example. Estimates for the oral bioavailability of midazolam are approximately 20% to 25%. Following the administration of midazolam 5 mg PO only, 1 mg to 1.5 mg survives degradation and first-pass metabolism to enter systemic circulation. Administered intramuscularly, the entire 5-mg dose will reach the systemic circulation, but the actual rate of absorption will vary. The delay in absorption following both PO and IM administration allows initially absorbed drug to distribute and eliminate before final portions enter the circulation. This does not allow a reliable prediction for peak serum levels and renders repeated doses perilous. Midazolam, 5 mg, administered by intravenous bolus will achieve almost instantaneous peak serum level. (This is mentioned here only for purposes of illustration. Such a dose should only be administered by those who have advanced training and intend to induce unconsciousness.) Stambaugh et al2 have provided data for meperidine serum levels that nicely illustrate these concepts and are presented in Figure 2.
Sublingual administration of a sedative such as triazolam introduces additional considerations. The drug is readily absorbed through nonkeratinized squamous epithelium in the floor of the mouth, and there is no loss due to gastric degradation. Furthermore, venous drainage does not enter the hepatic portal system for first-pass metabolism. However, one cannot predict the portion of a sublingual dose that is swallowed and is actually administered PO. Onset for some degree of sedation is more rapid due to the portion absorbed locally, but peak serum concentration will not occur until swallowed drug is absorbed in the intestines. Therefore, peak onset will be similar to that following conventional PO administration. Repeated “sublingual titration” becomes more and more problematic in terms of predicting the actual point at which peak concentration is achieved. This scientific principle has been confirmed impressively in clinical studies by Jackson et al3 and Pickrell et al4 (Figure 3).
The fact that repeated sublingual doses result in unpredictable peak serum concentrations introduces several significant clinical problems that are applicable to repeated PO dosing as well. The patient may remain stable in terms of vital functions while being supported and monitored, but the risk for deep sedation (unconsciousness) increases over time, and this must be avoided unless the dentist has received formal advanced training. Discharging a patient before peak serum levels have occurred is also a concern. The actual status of the patient at time for discharge is questionable since the serum concentration may actually be rising. Moreover, the relatively shorter elimination half-life for flumazenil renders the duration of benzodiazepine reversal inadequate.5 At this point in time we simply lack scientific evidence to support the safety of repeated oral or sublingual dosing. Furthermore, it goes without saying that regimens that combine various agents are even more perilous.
For purposes of sedation, IM injection offers few advantages over PO administration.6 Granted, the negative influences of gastric degradation and first pass metabolism are averted, but a fixed dose must still be predetermined and rates of absorption may vary to such an extent that effective serum concentrations are not attained. Likewise, repeated doses introduce identical concerns to those addressed above. Indications for this route are limited essentially to deep sedation regimens using ketamine or patients uncooperative with oral regimens.
Most of the concerns addressed above are obviated with intravenous (IV) administration. During IV sedation, neither the total dosage nor precise serum concentrations are crucial; small increments have been established that are safe and can be administered repeatedly until the intended level of sedation is achieved. Such accuracy provides a level of safety and efficacy for this method of drug administration that is unmatched by any other technique, with the exception of inhalation agents, so long as sufficient time is allowed between increments for the onset of the previous increment to peak. Although IV sedation may be more demanding technically, it is fallacious to consider it less safe than PO or IM regimens. It merely requires insertion of a catheter into a vein, a procedure taught to technical staff having no formal academic training.
Drug Circulation and Distribution
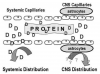
Once a drug is absorbed and bioavailable, it circulates throughout the bloodstream and distributes into most body tissues by diffusing through loosely joined capillary membranes. The rate for distribution is proportional to the relative perfusion (blood flow) of the tissue. Heart, kidney, liver, and brain are highly perfused and drug circulates most rapidly to these depots. The brain, however, introduces an additional consideration. The endothelium of capillaries within the central nervous system (CNS) is tightly joined and wrapped by astrocytes. This structural difference is described as the blood-brain barrier and requires the drug to have some degree of lipid solubility for it to leave the bloodstream and distribute into brain tissue (Figure 4).
While circulating within the bloodstream varying portions of drug may be loosely bound to plasma proteins. This is described as protein-binding and is generally of little importance because it is so highly dynamic. Consider for example a drug that demonstrates 50% protein binding, and a total of 100 drug molecules are circulating in the bloodstream. At this time 50 molecules will be bound to protein and 50 molecules will be circulating free and available to distribute. When 20 of these freely circulating molecules are distributed or eliminated there will be a total of 80 molecules remaining; 40 of these will be bound while 40 are free. This equilibration continues until all drug is eliminated. The dosage of the drug established by the manufacturer is predicated on this dynamic and the free drug portion. Concerns arise only when a patient’s medical status includes a reduction of circulating plasma protein. Severe liver and renal diseases are among the few examples where protein binding can become an issue and require a dosage reduction. The normal decline in plasma protein with age is not an issue and any concern has been overstated.7
Onset of Effect: Absorption and Distribution to Brain
Sedatives vary considerably in their lipid solubility, and this property enhances diffusion through cell membranes, which accounts for their differences in time for onset. For example, diazepam has far greater lipid solubility than lorazepam. Following PO administration, diazepam will absorb more rapidly through intestinal mucosa. Furthermore, this greater solubility speeds distribution of circulating drug through the blood-brain barrier and into the brain. The peak onset of diazepam following PO administration is 1 hour compared to 2 hours following a PO dose of lorazepam. The lower lipid solubility of lorazepam also explains why it has little use for intravenous titration of sedation. It distributes so slowly through the blood-brain barrier that an initial effect following an incremental dose requires several minutes, and the peak effect of an increment may take as long as 15 to 20 minutes.
Duration of Effect: Distribution versus Elimination
The duration of effect following administration of sedatives will obviously be related to the decline in serum concentration. However, there is a mistaken belief that drug elimination is the principal factor that accounts for this decline, and a drug having a longer elimination half-life will provide a longer duration of sedation. To correct this misconception we need to better appreciate the pharmacokinetics of distribution and elimination.
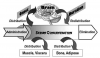
The so-called ‘‘two-compartment model’’ is conventionally used to describe the pharmacokinetics of drug distribution and elimination. When drug is administered it enters the bloodstream or central compartment. From this location it can undergo elimination or it can distribute into body tissues, the peripheral compartment. However, this peripheral compartment is very large and consists of all body tissues, each having varied degrees of volume and perfusion. For this reason a ‘‘three-compartment model’’ is more accurate because it divides the peripheral compartment into a ‘‘shallow’’ or highly perfused compartment and a vast ‘‘deep’’ compartment that is less perfused. This concept is illustrated in Figure 5.
Drug half-life (T1/2) is a general term representing the time required for plasma concentration to diminish by 50%. Unlike that following PO and IM administration, the decline in serum concentration following an IV bolus injection occurs in two major phases. The initial decline is rapid and is attributed to drug distribution, not elimination. The rate for this decline is designated distribution half-life (T1/2α) and is relevant only for drugs administered intravenously. (Following PO and IM administration, drug distribution proceeds simultaneously with absorption as illustrated in Figure 2.) Once distribution is completed, further decline in serum concentration becomes more gradual and is identical to that following PO and IM administration. This rate of decline is attributed to drug elimination and is designated elimination half-life (T1/2β). For example, the T1/2β for the drug illustrated in Figure 5 is approximately 3 hours, but its distribution half-life (T1/2β) is only 30 minutes.
Before the duration of drug effect is discussed any further, we need to clarify the significance of elimination half-life. This value provides two clinical correlates, neither of which relate to duration of sedation in clinical practice. The first of these is that a drug may be considered completely eliminated following four half-lives and, secondly, steady state drug concentrations can be achieved following four half-lives provided the drug is administered at a consistent dose and schedule.8 These principles are illustrated in Figure 6.
Although elimination half-life provides insight regarding the length of time a drug continues to circulate within the bloodstream, it does not identify the point at which the concentration falls below that required to sustain an adequate drug level in the target tissues. Following repeated administration, serum levels will become higher but a steady state will not occur unless dosing is consistent for a total of four half-lives. This consideration is more relevant for chronic drug therapy, not when drugs are administered for procedural sedation.
The duration of sedation is more dependent on drug distribution and redistribution because these processes generally proceed well before elimination comes into play. Revisit Figure 5 and imagine that a serum concentration of 35 ng/mL is required to provide adequate sedation. Notice that the process of distribution accounts for a drop below this level, not elimination. Therefore, the duration of sedation more closely correlates with distribution half-life (T1/2α) than elimination half-life (T1/2β).
Again consider diazepam (Valium) and lorazepam (Ativan) as examples. The elimination half-life for diazepam and its active metabolites ranges from 40 to 50 hours, while that for lorazepam is 15 to 20 hours. Yet the duration of sedation is significantly shorter for diazepam despite its longer elimination half-life. This is because diazepam has far greater lipid solubility. Even though adipose tissue is poorly perfused, highly lipid soluble drugs will distribute to this compartment more rapidly and account for a more rapid decline in the serum concentration. As this occurs, drug that initially distributed rapidly to brain will redistribute back into the blood stream reducing the degree of sedation. Therefore, high lipid solubility not only provides fast onset but accounts for shorter duration (Figure 7). This principle has been confirmed by Greenblatt et al following intravenous administration of diazepam and lorazepam9 and following PO administration of triazolam (Halcion) and zolpidem (Ambien).10
As serum levels rise following repeated doses, the duration of sedation may increase somewhat because tissue depots become more saturated and processes of elimination begin to come into play. During IV sedation using midazolam or diazepam, the duration of sedation following the first few increments may last only 10 to 15 minutes, which corresponds to the distribution half-lives for these drugs. During more lengthy appointments, however, subsequent IV increments may lead to progressively longer durations of sedation before additional increments are needed.
In summary, it is important to distinguish the relative significance of distribution versus elimination. While distribution and redistribution determine duration of sedation during treatment, drug elimination must be considered at discharge. Residual serum concentrations may not be profoundly sedative but can have an impact on subsequent psychomotor recovery. Furthermore, one cannot entertain the time for drug elimination until peak serum concentration has been achieved. This issue is of extreme importance following repeated PO or sublingual drug administration. Also, when considering drug elimination, attention must be given to any active metabolites of the parent drug. For example, normeperidine is an active metabolite of meperidine that acts as a CNS stimulant and has an elimination half-life of approximately 16 hours compared to only 2 to 3 hours for the parent drug.
Considerations for Continuous Infusions
Regimens administered by continuous infusion have become more popular among those practitioners with advanced training in deep sedation and general anesthesia. This is understandable because intermittent boluses result in oscillations in serum concentration above and below those desired for a procedure. An intermittent technique is acceptable for brief procedures lasting 20 to 40 minutes, but for more complex and lengthy procedures, a sustained and targeted level of moderate or deep sedation is far more attractive. The use of continuous infusions requires some additional understanding of pharmacokinetic principles.
As the duration of a continuous infusion is increased, the time for recovery can also be lengthened. This is because the drug concentration within peripheral compartments increases and mechanisms for elimination may become saturated. Either or both of these events can sustain serum concentrations because clearance of drug from the central compartment is prolonged. However, some drugs have unique properties that preclude this problem, and their duration is described using a concept called context-sensitive half time. This value addresses the time for serum concentrations to decline in the ‘‘context’’ of continuous infusions. Like drug “half-lives,” it represents a 50% reduction in serum concentration but commences at the time a continuous infusion is stopped. The subsequent decline in serum concentration reflects the combined influences of distribution and elimination. Some drugs such as propofol and remifentanil are well-suited for continuous infusions because they have short context-sensitive half-times that do not increase significantly during lengthy continuous infusions. While they accumulate in poorly perfused tissues over time, they are rapidly cleared as they slowly redistribute back into the bloodstream. Remifentanil is especially impressive because it is actually hydrolyzed by esterases within the tissues. Other drugs such as diazepam or fentanyl are inappropriate because they are cleared from the bloodstream more slowly. While midazolam is not cleared as rapidly as propofol, its rate of hepatic clearance is far greater than that for diazepam: 6 mL/kg/min to 11 mL/kg/min versus 0.2 mL/kg/min to 0.5 mL/kg/min.11 Midazolam is acceptable for continuous infusions but its context-sensitive half time increases over time and may not be attractive for lengthy procedures in outpatient practice. Representative context-sensitive half times are illustrated in Figure 8.
In summary it is important to appreciate the pharmacokinetic characteristics of all the medications we administer when providing any level of procedural sedation. Furthermore, it is essential to distinguish processes that influence the onset and duration for sedation from those that determine an adequate level of recovery for safe patient discharge.
About the Author
Dr. Becker is an Associate Director of Education, General Dental Practice Residency, at Miami Valley Hospital in Dayton, Ohio.
REFERENCES
1. Park GR, Gempeler F. Critical Care Management: Sedation and Analgesia. Philadelphia, Pa: WB Saunders Co; 1993:188.
2. Stambaugh JE, Wainer IW, Sanstead JK, Hemphill DM. The clinical pharmacology of meperidine—comparison of routes of administration. J Clin Pharmacol. 1976;16:245-256.
3. Jackson DL, Milgrom P, Heacox GA, Kharasch ED. Pharmacokinetics and clinical effects of multidose sublingual triazolam in healthy volunteers. J Clin Psychopharmacol. 2006;26:4-8.
4. Pickrell JE, Hosaka K, Jackson DL, et al. Expanded studies of the pharmacokinetics and clinical effects of multidose sublingual triazolam in healthy volunteers. J Clin Psychopharmacol. 2009;29:426-431.
5. Hosaka K, Jackson D, Pickrell JE, et al. Flumazenil reversal of sublingual triazolam: a randomized controlled clinical trial. J Am Dent Assoc. 2009;140:559-566.
6. Nicolson SC, Betts EK. Comparison of oral and intramuscular pre-anesthetic medication for pediatric inpatient surgery. Anesthesiology. 1989;71:8-10.
7. Grandison MK, Boudinot FD. Age-related changes in protein binding of drugs: implications for therapy. Clin Pharmacokinet. 2000;38:271-290.
8. Buxton ILO, Benet LZ. Pharmacokinetics: the dynamics of drug absorption, distribution, metabolism and elimination. In: Brunton LL, Chabner BA, Knollmann BC, eds. Goodman and Gilman’s The Pharmacological Basis of Therapeutics. 12th ed. New York, NY: McGraw-Hill Companies Inc; 2011.
9. Greenblatt DJ, Ehrenberg BL, Gunderman J, et al. Kinetic and dynamic study of intravenous lorazepam: comparison with intravenous diazepam. J Pharmacol Exp Ther. 1989;250:134-140.
10. Greenblatt DJ, Harmatz JS, von Moltke LL, et al. Comparative kinetics and response to the benzodiazepine agonists triazolam and zolpidem: evaluation of sex-dependent differences. J Pharmacol Exp Ther. 2000;293:435-443.
11. Reves JG, Glass PSA, Lubarsky DA, et al. Intravenous anesthetics. In: Miller RD, Eriksson LI, Fleisher LA, Wiener-Kroish JP, Young WL, eds. Miller’s Anesthesia. 7th ed. Philadelphia, Pa: Elsevier, Churchill Livingstone; 2009.
12. Hughes MA, Glass PS, Jacobs JR. Context-sensitive half-time in multicompartment pharmacokinetic models for intravenous anesthetic drugs. Anesthesiology. 1992;76:334-341.
13. Egan TD, Lemmens HJ, Fiset P, et al. The pharmacokinetics of the new short-acting opioid remifentanil (GI87084B) in healthy adult male volunteers. Anesthesiology. 1993;79:881-892.