You must be signed in to read the rest of this article.
Registration on CDEWorld is free. Sign up today!
Forgot your password? Click Here!
THE INFLAMMATORY PROCESS
What is inflammation? Isn't this the process that is supposed to be good for our bodies? Why does inflammation cause destruction of cells and tissues? As we learn more about the biological mechanisms of inflammation, it becomes clear that this process is more complicated than was once thought.
Inflammation is the body's response to cellular injury. Although there are harmful effects of inflammation, the fact remains that without this process, our bodies could not survive. Inflammation represents a protective response designed to rid the body of the initial cause of cell injury and the consequences of that injury. Cell injury may occur due to trauma, genetic defects, physical and chemical agents, tissue necrosis, foreign bodies, immune reactions and infections.
Inflammation is a local reactive change that involves the release of antibacterial agents from nearby cells that defend the host against infection. It also facilitates early tissue healing and repair. It contains, or "walls off," the infectious or injurious agent and serves as a defense mechanism that the body can use to restore itself to a normal morphological form and function.
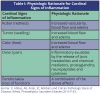
The inflammatory response consists of a vascular and a cellular reaction. These reactions are mediated by chemical factors derived from plasma proteins or cells. The classic signs of inflammation are redness, swelling, heat, pain and loss of function. The physiologic explanations for these signs appear in Table I. Other signs of inflammation include fever, leukocytosis or an increase in the number of circulating white blood cells, the presence of acute-phase proteins including C-reactive proteins (CRP), fibrinogen and serum amyloid A protein (SAA), and sepsis.
There are two types of inflammation: acute and chronic. Acute inflammation is characterized by a rapid onset and short duration. It manifests with exudation of fluid and plasma proteins, and emigration of leukocytes, most notably neutrophils. Examples of acute inflammation would be an ingrown toe nail, a sore throat from a cold virus, or the redness around a cut on the skin.
Chronic inflammation is of prolonged duration and manifests histologically by the presence of lymphocytes and macrophages and results in fibrosis and tissue necrosis. When inflammation continues for prolonged periods of time, it can be thought of as the healing process in overdrive, and deleterious changes can occur to localized tissues as well as the entire body. Chronic inflammation is also associated with excessive and inappropriate activation of the immune system leading to the development of autoimmune disease. Auto-antigens may cause an immune reaction resulting in chronic inflammation, an unregulated immune response against microbes leading to chronic inflammation, or an excessive response against common environmental substances causing allergic disease.1 Examples of chronic inflammation would be asthma, rheumatoid arthritis, gingivitis, periodontitis, and Crohn's disease.
In appreciating the inflammatory process, it is important to understand the role of chemical mediators in treating chronic inflammation. These are the substances that tend to direct the inflammatory response. These inflammatory mediators come from plasma proteins or cells including mast cells, platelets, neutrophils, and monocytes/macrophages. They are triggered by bacterial products or host proteins. Chemical mediators bind to specific receptors on target cells and can increase vascular permeability, vasodilation, and neutrophil chemotaxis, leukocyte recruitment and migration, stimulate smooth muscle contraction, have direct enzymatic activity, induce pain, or mediate oxidative damage. Most mediators are short-lived but cause harmful effects.1,2 Examples of chemical mediators include vasoactive amines (histamine, serotonin), arachidonic acids (prostaglandins, leukotrienes), cytokines (tumor necrosis factor and interleukin-1), complement proteins, and kinins.
Within a therapeutic context, the goal is to identify the underlying cause of inflammation to determine whether to promote or reduce inflammation. For example, in the event of infection, treatment is intended to increase the host response and eliminate the infection. Anti-infective medications, warm compresses, and gargling are examples that can be used to manage the infection and resultant inflammation. In the case of a traumatic injury, chronic inflammation can be reduced by using cold compresses and anti-inflammatory medications.1
INFLAMMATION AND ORAL HEALTH
The inflammatory process significantly affects the periodontium. Biofilm releases a variety of biologically active products as gram-positive and gram-negative bacteria colonize the tooth surface around the gingival margin and interproximal areas. These products include endotoxins, cytokines, and protein toxins.2 These molecules penetrate the gingival epithelium and initiate a host response that eventually results in gingivitis. Evidence of this can be seen clinically with changes in tissue color from pink to red, swelling, and bleeding upon probing.4 Because gingivitis is typically not painful, it may remain untreated for years. Worse, it may be viewed by practitioners as something that requires less concern than periodontitis. Nevertheless, chronic gingivitis that persists for years may provide the basis for greater concern for systemic health than a periodontitis condition that is more readily treated.
As the biofilm continues to proliferate, soluble compounds penetrate the sulcular epithelium. This, in turn, signals the gingival epithelium to produce chemical mediators including interleukin-1 beta (IL-1), prostaglandins, tumor necrosis factor alpha (TNF-a), and matrix metalloproteinases.5 These products recruit neutrophils to the area and influence chemotaxis, and can cause increased permeability of gingival vessels that permits plasma proteins to emigrate from the blood vessels into the tissue. As the inflammatory process progresses, additional mediators are produced, and more cell types are recruited to the area including neutrophils, T-cells, and monocytes. Continued inflammation results in signaling of fibroblasts and production of pro-inflammatory cytokines in the tissues. Antibodies specific to oral bacteria circulate in the peripheral blood. The acute-phase response becomes activated and CRP, fibrinogen, and complement are produced both by local cells and within the liver.6,7 These proteins may further exacerbate the local inflammatory response and may affect the initiation or progression of systemic disease (i.e., atherosclerosis).8,9 This process of chronic gingivitis is represented in Figure 1.
It is important to note that even though an individual may have established or chronic gingivitis, the condition is still reversible. Thorough dental hygiene debridement and regular home oral hygiene care could return the gingival tissues to a state of health. In some individuals when the inflammatory process continues and expands, the collagen of the periodontal ligament breaks down and bone resorption occurs, thus resulting in periodontitis. Individuals with periodontitis have the same increased levels of pro-inflammatory mediators as those with chronic gingivitis, including CRP, fibrinogen, and IL-1‚ and 6. Fortunately, when periodontal treatment is performed and clinical inflammation decreases, the serum levels of these inflammatory mediators also decrease.10
New research is examining additional ways to resolve inflammation beyond mechanical debridement and removal of the bacteria causing infection. Resolution of inflammation may also occur by receptor agonists that stop the pro-inflammatory activity of neutrophils and chemo-attractant leukotrienes while activating other cellular pathways producing anti-inflammatory and pro-resolution lipid mediators such as resolvins, lipoxins, and protectins.11 Madianos et al, suggest that the presence of a chronic inflammatory periodontal disease may be caused by too much pro-inflammatory signal or not enough pro-resolution signal. The host may respond to bacterial stimuli by over-secreting inflammatory mediators or by failing to produce sufficient pro-resolution signals.11 Although not yet supported by data in humans, animal studies of pro-resolution mediators have shown therapeutic benefits in periodontal disease including reducing bone destruction, reversing the levels of CRP and IL-1β levels, and promoting the regeneration of gingival and osseous tissues that were lost during active disease.12 Future treatment protocols for chronic gingivitis and periodontitis may include a combination of pro-resolution mediators and anti-inflammatory agents, such as aspirin and statins, in addition to mechanic removal of biofilm.
THE ORAL-SYSTEMIC RELATIONSHIP
Although periodontal diseases are well known as an oral problem, in the past decade, there has been a shift in perspective. Research has been focusing on the potential impact of periodontal diseases on systemic health. The relationship between periodontal inflammatory disease and systemic diseases such as cardiovascular disease, diabetes, respiratory disease, and adverse pregnancy outcomes has been closely investigated. Newer studies are beginning to investigate a relationship between periodontal pathogens and cancers. The basis for the biological mechanism of these relationships is emerging, and further study may lead to greater understanding of the connections and treatment approaches needed.
Cardiovascular disease (CVD) is characterized by the build-up of inflammatory plaques that may cause thromboses and eventual myocardial infarction. Atherosclerosis is the term used for the thickening and hardening of the arteries that is produced by this plaque build-up. It represents a chronic inflammatory response that causes injury to the endothelium of elastic and muscular arterial tissue. One of the hallmarks of the early atherosclerotic lesion is the presence of neutrophils followed by monocytes and lymphocytes.13 These leukocytes can affect the vascular endothelial lining and can cause oxidation of low-density lipoprotein (LDL) levels. Monocytes are induced to become macrophages, which take up modified lipoproteins and become lipid-laden "foam cells."14 The local inflammation is sustained by secreting chemical mediators, and the atherosclerotic lesion begins to bulge within the luminal wall. As this lesion progresses, the extracellular matrix is degraded by proteolytic enzymes and becomes susceptible to rupture. Thromboses can occur, occluding blood flow to the heart, which may eventually lead to infarction.
Since atherosclerosis is considered to be inflammatory in nature, identifying inflammatory markers that correlate with disease state is beneficial. One of the most recognized and consistent markers of systemic inflammation and poor cardiovascular prognosis is the acute-phase protein CRP.15,16 It is produced by the liver and released into the bloodstream. It is positively correlated to IL-6, activates complement, and accounts for LDL uptake by macrophages.17-19
It has been proposed that bacteria or viruses may directly infect atherosclerotic lesions contributing to the inflammatory process.20-22 Further, distant infections may increase systemic inflammation through the release of toxins or the leakage of chemical mediators into the circulation.23 It has been reported that studies of atheromatous lesions in the carotid arteries have found over 40% of atheromas contain antigens from periodontal pathogens including Porphyromonas gingivalis, Tannerella forsythensis, and Prevotella intermedia.24In addition, both in vitro and in vivo studies have shown that P. gingivalisand Streptococcus sanguiscan induce platelet aggregation, a component of atheroma and thrombus formation.25,26 This suggests a possible invasion of atheromas by oral pathogens as well as a possible contribution to their development. However, causality has yet to be established.
Animal model studies investigating the relationship between CVD and periodontal disease have demonstrated that clinically induced oral infection with P. gingivaliswill increase atheroma size and elevate CRP levels.27-31 Studies of humans reported a positive independent association between carotid intima-media thickness (IMT) and bacterial burden including P. gingivalis, Aggregatibacter actinomycetemcomitans, Treponema denticola, and T. forsythensis.32,33 Figure 2 represents the proposed connection between periodontal disease and atherosclerosis.
It is also thought that an autoimmune response may be involved in the development of atherosclerosis. Most humans have immune reactions against microbial heat-shock protein 60 (HSP60). Antibodies against bacterial versions of this protein may cross-react with human HSP60, causing an autoimmune response leading to endothelial cell damage, monocyte recruitment, elevated circulating lipids, and stimulating athersosclerosis.4,34-35
Paquette and Genco summarize numerous case-control and cohort studies that demonstrate levels of association between CVD and periodontal disease.36 Further, meta-analyses have been conducted on this association lending support to the existence of a connection between atherosclerosis and periodontal disease.37-39 However, a more recent review of existing research indicated there is a lack of causal evidence between periodontal disease and CVD.40 It is important to note that studies of this relationship have only examined associations and do not claim to suggest cause and effect. Ethical, methodological, and practical considerations of studying human subjects preclude the ability to answer questions about causality. Further study is needed to assess the effects of periodontal therapy on preventing the onset of CVD and slowing its progression.41 For example, a recent randomized controlled study compared a high dose of atorvastatin versus a low dose of the medication on periodontal disease. Findings showed that periodontal inflammation was significantly reduced in the high dose statin group. In addition, the study demonstrated that changes in periodontal inflammation correlated with changes in carotid inflammation, which may indicate a positive benefit for both carotid and extracardiac tissues such as the periodontium.42 This study may illuminate new treatment modalities for periodontal disease and CVD. As well, it provides additional evidence of the relationship between these diseases and their treatments.36
Diabetes mellitus is another systemic condition with oral inflammatory connections. One of the major complications of diabetes is periodontitis.43 While diabetes increases the probability of developing periodontal disease,43-45 periodontitis also increases the risk of poor glycemic control in people with diabetes when compared to those individuals with diabetes without periodontitis.46 Taylor et al reviewed research of periodontal disease and diabetes noting that evidence is emerging that suggests periodontal disease is associated with increased risk for diabetes complications, and may be associated with the development of gestational diabetes mellitus in addition to the development of type 2 diabetes mellitus.46 Fortunately, periodontal treatment can improve glycemic control by reducing the bacterial burden and the inflammatory response.48-50
There are several biological mechanisms proposed to explain the increased incidence and severity of periodontal disease in individuals with diabetes. Diabetes tends to increase susceptibility to infection- including oral infection-and the disease itself decreases the effectiveness of cells that kill bacteria. Another explanation is that inflammation is enhanced in those with diabetes. Research has demonstrated elevated levels of inflammatory mediators in the gingival crevicular fluid of periodontal pockets of poorly controlled patients with diabetes as compared to those without diabetes or those with diabetes who are well controlled. These patients had significant periodontal destruction with an equivalent bacterial challenge.45, 51, 52 In particular, the pro-inflammatory cytokine, TNF-a, plays a major role in this process. TNF-ahas a significant role in insulin resistance, the primary cause of type 2 diabetes. It is produced in large quantities by fat cells. Periodontitis has also been associated with increased levels of TNF-a. Elevated levels of TNF-amay lead to greater bone loss by killing cells that repair damaged connective tissue or bone and may exacerbate insulin resistance and worsen glycemic control.53-55 Other pro-inflammatory mediators produced in periodontal lesions, such as Il-1β, Il-6, prostaglandin E2, and thromboxane B2 may have important effects on glucose and lipid metabolism, insulin action, and diabetes and its complications.56-61
It has also been hypothesized that diabetes interferes with the capacity to form new bone after periodontal diseases have caused bone resorption. Graves et al studied genetically diabetic mice with type 2 diabetes and nondiabetic littermates by injecting them with P. gingivalis. The death of osteoblasts was measured, and results indicated that there was a higher and more prolonged rate of osteoblast cell death in the diabetic group. It was concluded that the capacity to repair a bony defect by producing new bone would be severely limited when osteoblasts died prematurely.62 Other animal studies have demonstrated increased periodontal bone loss linked to increased matrix metalloproteinase activity51 and increased production of receptor-activator of nuclear factor kappa B ligand (RANKL).63 Inhibiting these mechanisms showed significant reduction in bone loss.
Borgnakke summarized a review of available evidence related to the effect of periodontal infection on glucose levels including both randomized controlled clinical trials and non-randomized controlled trials.64 She notes that there are variations in the interventions, but most studies report a positive effect on blood glucose levels with nonsurgical periodontal therapy. The extent of the improvement in A1c level is similar to that achieved by adding a second oral antidiabetic medication to metformin. Therefore, nonsurgical periodontal therapy offers clinical significance for improving periodontal health and glycemic control. Borgnakke recommended additional well designed randomized controlled studies be performed to further evaluate this association.64
As with CVD and diabetes mellitus, there is a relationship between oral infection and respiratory disease. In particular, chronic obstructive pulmonary disease (COPD), community-acquired pneumonia (CAP) and nosocomial pneumonia have been associated with poor oral health.65-69 It is likely that oral biofilm serves as a reservoir of infection for respiratory bacteria. Specifically, Pseudomonas aeruginosa, Staphylococcus aureus, and enteric gram negative bacteria have been shown to colonize the teeth of patients admitted to hospitals or long-term care facilities. These bacteria may be released into saliva and then aspirated into the lower respiratory tract and lungs causing infection.5 Another vehicle by which bacteria from the oral cavity can be introduced into the respiratory system is intubation.
Inflammatory mediators, such as cytokines produced by the periodontium, may be another mechanism by which respiratory disease are associated with oral health. These mediators present in inflamed gingival tissues enter the gingival crevicular fluid and then the saliva. Once aspirated, these mediators can have proinflammatory effects in the lower airway.
Further, studies have demonstrated that periodontal diseases have been shown to increase the risk of adverse pregnancy outcomes such as premature birth, low birth weight and preeclampsia.70-76 Some studies have suggested that periodontal pathogens may travel from the gingival sulcus to the placenta and stimulate preterm birth. Specifically, Han and colleagues found that periodontal bacteria, including Fusobacterium nucleatum, entered the bloodstream from the oral cavity and directly affected the birth process.77 Lin et al found that bacterial complexes typically termed "orange" and "red" as well as low maternal IgG antibody response to periodontal bacteria during pregnancy was associated with an increased risk for preterm delivery, and that higher levels of these periodontal pathogens were measured antepartum in the preterm deliveries compared with term deliveries.78
Studies examining the effect of the treatment of periodontal disease with debridement on the prevention of preterm birth have been conducted, yet remain inconclusive. Two meta-analyses have found that periodontal disease increases the risk of premature birth and that treatment decreases that risk.79-80 However, a large clinical trial failed to show obstetric benefits in relation to periodontal care.81 Another multicenter, randomized controlled study of pregnant women with periodontal disease who received periodontal therapy and standard obstetric care did not show findings that altered the rates of preterm birth or fetal growth restriction.82 Thus, future study is needed to determine whether periodontal disease can influence reverses in preterm birth or other adverse pregnancy events.79 As Barros and Offenbacher note, studies have shown that periodontal treatment can be performed safely to pregnant women, and maternal periodontal health in itself is an important outcome regardless of other pregnancy outcomes. Further, how periodontal organisms interact with each other or separately to affect pregnancy complications needs to be better understood. Whether these processes reflect an underlying genetic predisposition due to a hyperinflammatory response or some other factors are occurring remains to be discovered.83
A summary of the biological mechanisms by which gingival inflammation affects systemic health appears in Figure 3.
TRANSLATING SCIENCE TO PRACTICE
As often occurs, research provides many answers and drives more questions. Despite the knowledge obtained through science, it is incumbent upon clinical practitioners to translate the evidence into practical use.
• What does the above information mean for clinical practice?
• Does it offer opportunities for changing oral health interventions?
• How can we use this information to answer patients' questions when they inquire about the relationship between periodontal disease and general health after reading articles in magazines or seeing news stories on the television?
Understanding the association between oral health and systemic health does provide opportunities for the dental team to reframe their protocols. The process begins by practicing oral medicine. First, comprehensive medical assessment is needed for each patient. A review of systems and vital signs evaluation should be part of that assessment process. During these assessments, identifying risk factors for specific systemic diseases is important, including age (over 40 years), hypertension, dyslipidemia, smoking, obesity/overweight, CVD, diabetes or symptoms of diabetes, and women who are pregnant and have poor oral hygiene. Conferring and collaborating with medical professionals sends the message that a concerted effort is needed to improve both oral and general health.
Comprehensive oral assessment is equally important. This may include thorough head and neck examination, radiographic imaging (if clinically indicated), periodontal probing, bacterial monitoring of periodontal and carious pathogens, genetic testing for periodontal disease, or other diagnostic tools that seem appropriate to each individual need.
As these assessments are being performed, risk factors for oral and systemic diseases are being noted and explained to the patient. It is essential that the patient understand that the purpose of these assessments is to prevent problems from occurring or treat them as readily as possible. Patients are well acquainted with the idea that physicians prefer to treat a stroke or a heart attack before it occurs by identifying possible risk factors and trying to reduce them. They are used to attending a medical appointment and having certain assessments performed. They are even used to requesting certain tests or procedures based on their own education and experience. Thus, it is time that we incorporate these approaches into practice. Our patients need to become accustomed to the same comprehensive assessment process. Clinicians can then put together a picture for the patient that incorporates oral and systemic risk factor findings, and discuss how their chronic gingivitis or periodontitis condition may influence their overall health.
Once risks have been identified, those that can be modified are incorporated into the dental hygiene treatment plan and patient education process. Just as a physician will recommend a patient lose weight or prescribe an antihypertensive agent, the dental healthcare professional may make recommendations for the patient to begin a smoking cessation program, use specific preventive oral care products, monitor their blood sugar regularly or complete a nutrition counseling program, in addition to having debridement of biofilm and calculus. Patients may place greater value on their oral health care if greater emphasis is placed on the patient's total health, risk factor assessment, and risk factor modification.
In addition, once risk factors have been identified and appropriate treatment planned, it is important to be prepared to answer questions about medications and products that have anti-inflammatory and/or antibacterial properties. Medications used to treat systemic diseases, such as autoimmune diseases with an inflammatory component, may be investigated for their role in treating chronic inflammation related to gingivitis and periodontitis. Medications that focus on switching from the production of pro-inflammatory molecules to pro-resolving molecules have been investigated.84-86 Specialized pro-resolving mediators such as resolvins, protectins, maresins, and annexin A1 may be key to providing alternative ways of managing chronic inflammation.80-82 It is conceivable that combining an aspirin and/or statin regimen with a pro-resolving medication may be a treatment regimen of the future for managing periodontal disease.
A current anti-inflammatory medication that has been shown to be effective for the treatment of periodontitis is low dose doxycycline hyclate (Periostat®). Periostat inhibits the collagenase activity by neutrophils, thus preventing the degradation of connective tissue and bone loss. Therefore, it is beneficial as part of host modulation therapy. It is administered twice daily at a dosage of 20 mg. Periostat is an antibiotic; however, the dose is too low to produce antibacterial effects. Studies have demonstrated that Periostat improves the effectiveness of routine scaling and root planing and that the progression of periodontitis is decreased.87
Optimal preventive education programs should include discussion of twice-daily brushing, flossing, and use of a chemotherapeutic mouth rinse to reduce bacterial plaque and susceptibility to gingivitis.88 Products recommended should be those that have been well-researched and demonstrated safety and efficacy. For example, Peridex® and Listerine® Antiseptic Mouthrinse are the only two chemotherapeutic mouth rinses that have been approved by the American Dental Association Council on Scientific Affairs. Their effectiveness has been well established. Similarly, a dentifrice containing triclosan/copolymer (Colgate Total® Toothpaste) has been shown to be effective in reducing plaque and gingivitis, controlling bacterial infection, and preventing or slowing the progression of periodontal disease.89 In addition, triclosan has been shown to possess potent anti-inflammatory properties. In vitro studies have demonstrated that triclosan has inhibited IL-1 stimulated prostaglandin production in human gingival fibroblast cells, inhibited the production of IL-1 by fibroblasts stimulated with TNF-a,and has inhibited the production of collagenases by human bone cells and fibroblasts stimulated with IL-1 and TNF-a.90, 91 In addition, the safety of this product has been well studied and established.92 The antibacterial and anti-inflammatory properties of triclosan are reasons to recommend toothpaste containing this ingredient for patients with periodontal diseases as well as for those whose systemic health has been compromised.
SUMMARY
Research suggests that there is an interrelationship between oral infection, inflammation and systemic health. Patients, dental healthcare professionals and other health care providers should be aware of the consistent relationships between oral inflammation and systemic diseases. They should value the need to modify assessment, prevention, and treatment protocols to improve the oral health as well as total health of the patients they treat in the office each day.
REFERENCES
1. Kumar V, Abbas AK, Aster JC. Robbins and Cotran Pathologic Basis of Disease, 9th ed. 2015. Philadelphia, PA: Elsevier Saunders, p. 69-112.
2. Mariotti A. A primer on inflammation. Compend Cont Educ Dent 2004; 25 (7) (Suppl 1):7-15.
3. Kornman KS, Page RC, Tonetti MS. The host response to the microbial challenge in periodontitis: assembling the players. Periodontol 2000 1997; 14:33-53.
4. Armitage GC. Diagnosis of periodontal diseases. J Periodontol. 2003; 74; 1237-47.
5. Scannapieco FA: Periodontal inflammation: from gingivitis to systemic disease? Compend Cont Educ Dent. 2004; 25 (7) (Suppl 1): 16-25.
6. Ebersole JL, Machen RL, Steffen MJ, et al. Systemic acute-phase reactants, C-reactive protein and haptoglobin, in adult periodontitis. Clin Exp Immunol. 1997; 107: 347-52.
7. Loos BG, Craandijk J, Hoek FL, et al. Elevation of systemic markers related to cardiovascular disease in the peripheral blood of periodontitis patients. J Periodontol. 2000; 71: 1528-34.
8. Danesh J, Collins R, Appleby P, et al. Association of fibrinogen, C-reactive protein, albumin, or leukocyte count with coronary heart disease: metaanalyses of prospective studies. J Am Med Assoc. 1998; 279: 1477-82.
9. Ridker PM, Buring JE, Shih J, et al. Prospective study of C-reactive protein and the risk of future cardiovascular events among apparently healthy women. Circulation. 1998;98:731-33.
10. D'Aiuto F, Parkar M, Andreou G, et al. Periodontitis and systemic inflammation: control of the local infection is associated with a reduction in serum inflammatory markers. J Dent Res. 2004; 83:156-60.
11. Madianos PN, Bobetsis YA, Van Dyke TE. Infection and inflammation. In Genco RJ, Williams RC. Periodontal disease and overall health: A clinician's guide, 2nd ed. 2014. Yardley: Professional Audience Communications, Inc. p. 30-48.
12. Hasturk H, Kantarci A, Ohira T, et al. RvE1 protects from local inflammation and osteoclast-mediated bone destruction in periodontitis. FASEB J. 2006;20:401-403.
13. Schwartz CJ, Valente AJ, Sprague EA, et al. The pathogenesis of atherosclerosis: an overview. Clin Cardiol. 1991; 14 (2 suppl 1): 11-6.
14. Paigen B, Morrow A, Holmes PA, et al. Quantitative assessment of atherosclerotic lesions in mice. Atherosclerosis. 1987; 68: 231-40.
15. Ridker PM, Hennekens CH, Buring JE, et al. C-reactive protein and other markers of inflammation in the prediction of cardiovascular disease in women. N Engl J Med. 2000; 342: 836-43.
16. Liuzzo G, Biasucci LM, Gallimore JR, et al. The prognostic value of C-reactive protein and serum amyloid A protein in severe unstable angina. N Engl J Med. 1994; 331: 417-24.
17. Ruderman NB, Williamson JR, Brownlee M. Glucose and diabetic vascular disease. FASEB J. 1992; 6: 2905-14.
18. Bhakdi S, Torzewski M, Klouche M, et al. Complement and atherogenesis: binding of CRP to degraded, nonoxidized LDL enhances complement activation. Arterioscler Thromb Vasc Biol. 1999; 19: 2348-54.
19. Zwaka TP, Hombach V, Torzewski J. C-reactive protein-mediated low density lipoprotein uptake by macrophages: implications for atherosclerosis. Circulation. 2001; 103: 1194-7.
20. Deshpande RG, Khan MB, Genco CA. Invasion of aortic and heart endothelial cells by Porphyromonos gingivalis. Infect Immun. 1998;66:5337-5343.
21. Chiu B. Multiple infections in carotid atherosclerotic plaques. Am Heart J. 1999;138:534-536.
22. Haraszthy VI, Zambon JJ, Trevisan M, Zeid M, Genco RJ. Identification of periodontal pathogens in atheromatous plaques. J Periodontol. 2000;71:1554-1560.
23. Epstein SE. The multiple mechanisms by which infection may contribute to atherosclerosis development and course. Circ Res. 2002; 90: 2-4.
24. Haraszthy VI, Zambon JJ, Trevisan M, et al. Identification of periodontal pathogens in atheromatous plaques. J Periodontol. 2000; 71 (10): 1554-60.
25. Herzberg MC, Meyer MW: Effects of oral flora on platelets: possible consequences in cardiovascular disease. J Periodontol. 1996; 67 (10Suppl): 1138-42.
26. Herzberg MC, Myer MW. Dental plaque, platelets and cardiovascular disease. Ann Periodontol 1998;3:152-160.
27. Paquette DW. The periodontal-cardiovascular link. Compend Cont Educ Dent. 2004; 25 (9):681-92.
28. Chi H, Messas E, Levine RA, Graves DT, Amar A. Interleukin-1 receptor signaling mediates atherosclerosis associated with bacterial exposure and/or a high-fat diet in a murine apoliopoprotein E heterozygote model: pharmacotherapeutic implications. Circulation. 2004;110:1678-1685.
29. Gibson FC 3rd, Hong C, Chou HH, Yumoto H, Chen J, Lien E, Wong J, Genco CA. Innate immune recognition of invasive bacteria accelerates atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2004;109:2801-2806.
30. Lalla E, Lamster IB, Hofmann MA, Bucciarelli L, Jerud AP, Tucker S, Lu Y, Papapanou PN, Schmidt AM. Oral infection with a periodontal pathogen accelerates early atherosclerosis in apolipoprotein E-null mice. Arterioscler Thromb Vasc Biol. 2003;23:1405-1411.
31. Li L, Messas E, Batista El, Jr., Levine RA, Amar S. Porphyromonos gingivalis infection accelerates the progression of atherosclerosis in a heterozygous apolipoprotein E-deficient murine model. Circulation. 2002;105:861-867.
32. Desvarieux M, Demmer RT, Rundek T, et al. Periodontal microbiota and carotid intima-media thickness: the oral infections and vascular disease epidemiology study (INVEST). Circulation. 2005; 111 (5): 576-82.
33. Engebretson SP, Lamster IB, Elkind MS, Rundek T, et al. Radiographic measures of chronic periodontitis and carotid artery plaque. Stroke. 2005;36:561-566.
34. Hinode D, Nakamura R, Gernier D, Mayrand D. Cross-reactivity of specific antibodies directed to heat shock proteins from periodontopathogenic bacteria and of human origin. Oral Microbiol Immunol. 1998;13:55-58.
35. Simes TJ, Lernmark A, Mancl LA, Schifferle RE, Page RC, Persson GR. Serum IgG to heat shock proteins and Porphyromonos gingivalis antigens in diabetic patients with periodontitis. J Clin Periodontol. 2002;29:551-562.
36. Paquette DW, Genco RJ. Association between periodontal disease and atheromatous disease. In Genco RJ, Williams RC. Periodontal disease and overall health: A clinician's guide, 2nd.ed. 2014. Yardley: Professional Audience Communications, Inc. p.131-151.
37. Meurman JH, Sanz M, Janket SJ. Oral health, atherosclerosis and cardiovascular disease. Crit Rev Oral Biol Med. 2004;15:403-413.
38. Khader YS, Albashaireh, ZS, Alomari MA. Periodontal diseases and the risk of coronary heart and cerebrovascular diseases: a meta-analysis. J Periodontol. 2004;75:1046-1053.
39. Vettore MV. Periodontal disease and cardiovascular disease. Evid Based Dent. 2004:5:69.
40. Lockhart PB, Bolger AF, Papapanou PN, Osinbowale O, et al. Periodontal disease and atherosclerotic vascular disease: does the evidence support an independent association?: A scientific statement from the American Heart Association. Circulation. 2012;125:1-25. Available at: http://circ.ahajournals.org. Accessed June 17, 2013.
41. Papapanou PN, Trevisan M. Periodontitis and atherosclerotic vascular disease: what we know and why it is important. JADA. 2012;143(8):826-828.
42. Subramanian S, Emami H, Vucic E, et al. High-dose atorvastatin reduced periodontal inflammation. A novel pleiotropic effect of statins. J Am Coll Cardiol. 2013;62:2382-91.
43. Löe H. Periodontal disease. The sixth complication of diabetes mellitus. Diabetes Care. 1993; 16: 329-34.
44. Nishimura F, Takahshi K, Kurihara M, et al. Periodontal disease as a complication of diabetes mellitus. Ann Periodontol. 1998;3:20-29.
45. Ryan ME, Carnu A, Kamer A. The influence of diabetes on the periodontal tissues. J Am Dent Assoc. 2003; 134: 34S-40S.
46. Taylor GW, Burt BA, Becker MP, et al. Severe periodontitis and risk for poor glycemic control in patients with non-insulin-dependent diabetes mellitus. J Periodontol. 1996; 67 (10 Suppl): 1085-93.
47. Taylor GW, Borgnakke WS, Graves DT. Association between periodontal diseases and diabetes mellitus. In Genco RJ, Williams RC. Periodontal disease and overall health: A clinician's guide. 2010. Yardley: Professional Audience Communications, Inc. p.83-104.
48. Grossi SG, Skrepcinski FB, DeCaro T, et al. Treatment of periodontal disease in diabetics reduces glycated hemoglobin. J Periodontol. 1997; 68: 713-9.
49. Miller LS, Manwell MA, Newbold D, et al. The relationship between reduction in periodontal inflammation and diabetes control: a report of 9 cases. J Periodontol. 1992; 63: 843-8.
50. Mealey DL, Rethman MP: Periodontal disease and diabetes mellitus. Bidirectional relationship. Dent Today. 2003; 22: 107-13.
51. Ryan ME, Ramamurthy NS, Corsa T, Golub LM. MMP mediated events in diabetes. Ann NY Acad Sci. 1999; 878: 331-4.
52. Ryan ME, Usman A, Ramamurthy NS, et al. Excessive matrix metalloproteinase activity in diabetes: inhibition by tetracycline analogues with zinc reactivity. Curr Med Chem. 2001; 8 (3): 305-16.
53. Salvi GE, Yalda B, Collins JG, et al. Inflammatory mediator response as a potential risk marker for periodontal diseases in insulin-dependent diabetes mellitus patients. J Periodontol. 1997; 68: 127-35.
54. Lalla E, Lamster IB, Feit M, et al. Blockade of RAGE suppresses periodontitis-associated bone loss in diabetic mice. J Clin Invest. 2000; 105: 1117-24.
55. Grossi SG, Genco RJ. Periodontal disease and diabetes mellitus: a two-way relationship. Ann Periodontol. 1998; 3: 51-61.
56. King GL. The role of inflammatory cytokines in diabetes and its complications. J Periodontol. 2008;79:1537-1534.
57. Feingold KR, Soued M, Serio MK, Moser AH, et al. Multiple cytokines stimulate hepatic lipid synthesis in vivo. Endocrinology. 1989;125:267-274.
58. Ling PR, Istfan NW, Colon E, Bistrian BR. Differential effects of interleukin-1 receptor antagonist in cytokine-and endotoxin-treated rats. Am J Physiol. 1995;268:E255-E261.
59. Feingold KR, Grunfeld C. Role of cytokines in inducing hyperlipidemia. Diabetes. 1992;41 (Suppl):97-101.
60. Grunfeld C, Soued M, Adi S Moser AH, et al. Evidence for two classes of cytokines that stimulate hepatic lipogenesis: relationships among tumor necrosis factor, interleukin-1 and interferon-alpha. Endocrinology. 1990;127:46-54.
61. Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. 2006;116:1793-1801.
62. Graves DT, Al-Mashat H, Liu, RL. Evidence that diabetes mellitus aggravates periodontal diseases and modifies the response to an oral pathogen in animal models. Compend Cont Educ Dent. 2004; 25 (7) (Suppl 1): 38-45.
63. Mahamed DA, Marleau A, Alnaeeli M, Singh B, et al. G(-) anaerobes-reactive CD4+ T-cells trigger RANKL-mediated enhanced alveolar bone loss in diabetic NOD mice. Diabetes. 2005;54:1477-1486.
64. Borgnakke WS. Hyperglycemia/diabetes mellitus and periodontal infection adversely affect each other. In Genco RJ, Williams RC. Periodontal disease and overall health: A clinician's guide, 2nd. ed. 2014. Yardley: Professional Audience Communications, Inc. p. 99-122.
65. Scannapieco FA. Role of oral bacteria in respiratory infection. J Periodontol. 1999; 70: 793-802.
66. Scannapieco FA, Bush RM, Paju S. Associations between periodontal disease and risk for nosocomial bacterial pneumonia and chronic obstructive pulmonary disease: a systematic review. Ann Periodontol. 2003; 8: 54-69.
67. Hayes C, Sparrow D, Cohen M, et al. The association between alveolar bone loss and pulmonary function: the VA Dental Longitudinal Study. Ann Periodontol. 1998; 3: 257-61.
68. Scannapieco FA, Ho AW. Potential associations between chronic respiratory disease and periodontal disease: analysis of National Health and Nutrition Examination Survey III. J Periodontol. 2001; 72: 50-6.
69. Scannapieco FA, Mylotte JM. Oral health and diseases of the respiratory tract. In Genco RJ, Williams RC. Periodontal disease and overall health: A clinician's guide, 2nd ed. 2014. Yardley: Professional Audience Communications, Inc. p 176-192.
70. Offenbacher S, Katz V, Fertik G, et al. Periodontal infection as a possible risk factor for preterm low birth weight. J Periodontol. 1996; 67 (10 suppl): 1103-13.
71. Jeffcoat MK, Guers NC, Reddy MS, et al. Periodontal infection and preterm birth: results of a prospective study. J Am Dent Assoc. 2001; 132: 875-80.
72. Scannapieco FA, Bush RP, Paju S. Periodontal disease as a risk factor for adverse pregnancy outcomes. A systematic review. Ann Periodontol. 2003; 8: 70-8.
73. Cadakci V, Canakci CF, Yildirim A, Ingec M, et al. Periodontal disease increases the risk of severe preeclampsia among pregnant women. J Clin Periodontol. 2007;34:639- 645.
74. Boggess KA, Lieff S, Murtha AP, Moss K, et al. Maternal periodontal disease is associated with an increased risk for preeclampsia. Obstet Gynecol. 2003;101:227-231.
75. Barak S, Oettinger-Barak O, Machtei EE, Sprecher H, Ohel G. Evidence of periopathogenic microorganisms in placentas of women with preeclampsia. J Periodontol. 2007;78:670-676.
76. Contreras A, Herrera JA, Soto JE, Arce RM, et al. Periodontitits is associated with preeclampsia in pregnant women. J Periodontol. 2006;77:182-188.
77. Han YW, Redline RW, Li M, et al. Fusobacterium nucleatum induces premature and term stillbirth in pregnant mice: implication of oral bacteria in preterm birth. Infect Immun. 2004; 72: 2272-9.
78. Lin D, Moss K, Beck JD, et al. Persistently high levels of periodontal pathogens associated with preterm pregnancy outcome. J Periodontol. 2007;78(5):833-41.
79. LόpezNJ, Da Silva I, Ipinza J, Gutiérrez J. Periodontal therapy reduces the rate of preterm low birth weight in women with pregnancy-associated gingivitis. J Periodontol. 2005;76(11 Suppl):2144-2153.
80. Polyzos NP, Polyzos IP, Mauri D, Tzioras S, et al. Effect of periodontal disease treatment during pregnancy on preterm birth incidence: a metaanalysis of randomized trials. Am J Obstet Gynecol. 2009;200:225-232.
81. Michalowicz BS, Hodges JS, DiAngelis AJ, Lupo VR, et al. OPT study. Treatment of periodontal disease and the risk of preterm birth. N Engl J Med. 2006;355:1885-1894.
82. Offenbacher S, Beck JD, Jared HL, et al. Maternal oral therapy to reduce obstetric risk (MOTOR) investigators. Effects of periodontal therapy on rate of preterm delivery: a randomized control trial. Obstet Gynecol. 2009:114(3):551-9.
83. Barros S Offenbacher S. Adverse pregnancy effects. In Genco RJ, Williams RC. Periodontal disease and overall health: A clinician's guide, 2nd. Ed. 2014. Yardley: Professional Audience Communications, Inc. p 152-175.
84. Serhan CN. Systems approach to inflammation resolution: identification of novel anti-inflammatory and pro-resolving mediators. J Thromb Haemost. 2009;7 (Suppl)1:44-48.
85. Lee HN, Surh YJ. Therapeutic potential of resolvins in prevention and treatment of inflammatory disorders. Biochem Pharmacol. 2012;84(10):1340-1350.
86. Alessandri AL, Sousa LP, Lucas CD, Rossie AG, et al. Resolution of inflammation: mechanisms and opportunity for drug development. Pharmacol Ther. 2013.doi:10.1016/j.pharmthera.2013.04.006. [Abstract].
87. Panagakos FS. Inflammation: its role in health and its mediation by chemotherapeutic agents. Continuing Education for the Healthcare Professional (CEHP), distributed by Sullivan-Schein, a Henry Schein Company, course reference #05AS2906B, 2005.
88. Gurenlian JR. Diabetes mellitus: strategies for providing comprehensive care. Continuing Education for the Healthcare Professional (CEHP), distributed by Sullivan-Schein, a Henry Schein Company, course reference # 05AS2904, 2005.
89. Gaffar A, Scherl D, Afflitto J, Coleman EJ. The effect of triclosan on mediators of gingival inflammation. J Clin Periodontol. 1995; 22(6): 480-4.
90. Xu T, Deshmukh M, Barnes VM, et al. Effectiveness of a triclosan/copolymer dentifrice on microbiological and inflammatory parameters. Compend Cont Educ Dent. 2004; 25 (7) (Suppl 1): 46-53.
91. DeVizio W, Davies R. Rationale for the daily use of a dentifrice containing triclosan in the maintenance of oral health. Compend Cont Educ Dent. 2004; 25 (7) (Suppl 1): 54-7.
92. Spolarich AE. Triclosan safety exposed. October 2014. Colgate Oral Health Network. Available at www.colgateoralhealthnetwork.com/article/triclosan-safety-exposed/. Accessed February 12, 2017.