You must be signed in to read the rest of this article.
Registration on CDEWorld is free. Sign up today!
Forgot your password? Click Here!
SEDATION VERSUS GENERAL ANESTHESIA
Sedation is a continuum that proceeds from minimal to deep in a dose-response manner. This continuum can be divided into levels having characteristics that have been used to design several subjective sedation scales. Table 1 summarizes a suggested clinical scale derived from both the Ramsay Scale commonly used in critical care medicine2 and the Observer’s Assessment of Alertness/Sedation Scale.3
While levels of sedation follow a continuum, it is significant that general anesthesia is distinct. Any reference to the dated Guedel stages and planes of general anesthesia is no longer applicable. The Guedel stages pertained only to ether anesthesia and are not produced reliably by any of the intravenous or inhalation agents used currently in general anesthesia practice. Today, the term general anesthesia reflects a state in which three principal components are fulfilled: (a) unconsciousness and amnesia (hypnosis), (b) complete analgesia (areflexia), and (c) immobilization (muscle relaxation).4,5 Each component is accomplished by drug actions at distinct locations within the central nervous system (CNS), and only the inhalation anesthetics are capable of providing all three components.5 The term general anesthetic is used spuriously when describing potent sedative-hypnotics such as propofol and methohexital. Unconsciousness (deep sedation) alone does not define general anesthesia. For example, propofol provides hypnosis but a surgical stimulus typically evokes autonomic and somatic reflexes that confirm the absence of significant analgesia and immobilization, unless local anesthesia is also present.
While levels of sedation progress in a dose-response continuum, it is not always possible to predict precisely how an individual patient will respond to a particular dose. Hence, practitioners intending to produce a given level of sedation should be able to rescue patients whose level of sedation becomes deeper than initially intended. For example, individuals administering moderate sedation (conscious sedation) should be able to rescue patients who enter a state of deep sedation (unconsciousness). Regardless of the particular drugs that may be used to provide sedation, or general anesthesia for that matter, it is important to appreciate the general influences of various levels of CNS depression on body functions. These are summarized in Table 2 as provided by the American Society of Anesthesiologists in guidelines published for non-anesthesiologists.6
DRUG ACTIONS AND EFFECTS
The action of a drug is the biologic mechanism by which the drug interacts with tissues to produce a change or effect. Drug effects are conventionally categorized as primary when intended, but drugs always produce additional effects as well. These secondary effects are described arbitrarily as side, adverse, or toxic effects depending on their severity. For example, opioid effects include analgesia, sedation, and respiratory depression. While sedation may be viewed as a side effect of opioids when prescribed for pain, it is a primary effect when used to manage apprehensive patients.
When describing drug effects, the terms potency and efficacy are often used improperly. Efficacy refers to the magnitude of the clinical effect that is produced by a drug’s action. Potency refers to the dose that will produce a specific intensity of effect. The significance of drug potency is commonly overstated. Drugs having identical mechanisms of action generally produce comparable efficacy. Their dosages (potency) may differ, but equipotent doses will result in the same intensity of effect. This concept is illustrated in Figure 1.
There are several mechanisms of action by which drugs can calm and sedate the apprehensive patient. The most effective mechanisms enhance chloride ion influx through channels modulated by gamma aminobutyric acid (GABA). Drug mechanisms that target additional neurotransmitters are generally less effective when used alone but provide a synergistic influence when added to a regimen. In most cases, drugs used for sedation act on specific receptors as either agonists or antagonists. An agonist binds to a receptor and initiates or activates a biochemical event leading to the effect. Frequently they mimic an endogenous ligand that normally modulates the specific receptor. An antagonist binds to a receptor but is incapable of initiating a response. However, its presence on the receptor blocks any action by the endogenous ligand or other agonists for the receptor. For example, opioid agonists mimic the action of endorphins on mu opioid receptors, while the antagonist, naloxone, can reverse these effects. General drug mechanisms are summarized in Table 3 and will be further explained in subsequent sections of this article.
A large number of sedatives, opioids, and adjunctive agents can be used for procedural sedation. The general pharmacology and characteristics for each drug class should be thoroughly understood; drugs within each class are similar with only minor differences that make each agent unique, eg, dosage and patterns of clearance.
To select the drug or combination of drugs to be employed, one first selects the class whose pharmacologic profile will, in general, fulfill one or more of the objectives to be attained. Examples might include anxiolysis, sedation, amnesia, analgesia, antiemetic, or antisecretory (anticholinergic). For instance, an apprehensive patient who becomes easily nauseated might very well be managed using a combination of a benzodiazepine and an antihistamine, avoiding the nauseating potential of opioids. The specific agent selected from each of these classes will depend on subtle differences the provider deems an advantage. Duration of effect, expense, and pattern of clearance are likely parameters to be considered.
In order to simplify drug selection, the number of drug classes useful for the production of moderate to deep sedation will be restricted to four: sedative-anxiolytics, opioids, antihistamines, and nitrous oxide. When a multiple-drug regimen is employed during intravenous techniques, the sequence of drug administration is, to a large extent, a matter of personal preference. However, because sedative-anxiolytics are most accurately titrated to the desired effect, and because they alone may produce adequate sedation in many patients, it seems logical to commence with one of these agents. An antihistamine, opioid, or nitrous oxide can then be titrated as needed. For enteral techniques, titration cannot be recommended, but a single dose of a sedative-anxiolytic can be followed by titration of nitrous oxide if necessary.
The titrated end-point for intravenous or enteral nitrous oxide moderate sedation is reached when the patient is noted to be calm and relaxed. Eyelids typically droop, speech becomes slurred, and patients often lose train of thought. For providers with advanced training who intend deep sedation, the endpoint is obviously unconsciousness.
BENZODIAZEPINES
Benzodiazepines are the conventional drugs of choice for most procedural sedation regimens, regardless of intended depth or route of administration. Benzodiazepines have a high therapeutic index and a relatively shallow dose-response curve. A high therapeutic index means that the dose of the agent required to produce desired effects is considerably less than that required to produce adverse effects. Furthermore, the shallow dose-response indicates that a dose required to produce minimal to moderate sedation is well below that required to produce hypnosis (unconsciousness) in all but the most sensitive patients, eg, the elderly, debilitated, or those with obstructive sleep apnea.
Actions and Effects
The therapeutic effects of benzodiazepines are attributed to their ability to potentiate the inhibitory influences of GABA. This endogenous neurotransmitter and its receptor complex are among the most researched and understood targets for drugs that depress the CNS.
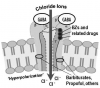
GABA is the principal inhibitory neurotransmitter in the mammalian brain. There are three major subtypes of GABA receptors designated GABAA, GABAB, and GABAC, but only the GABAA receptor regulates chloride ion channels within neurons of the brain and is the target of many sedatives and general anesthetics.7 The GABAA receptor is actually a complex protein that forms chloride ion channels (chloride ionophores) found in neuronal cell membranes. Upon its neuronal release, GABA binds to this receptor on adjacent neurons, opening the channel for chloride ions to enter the cell. The influx of negative ions hyperpolarizes the neuron, rendering it less responsive to excitatory signals.
The protein containing the GABAA receptor is vast and composed of many subunits of which alpha, beta, and gamma subunits are most defined. They are further divided into subunit families designated by numerical subscripts, eg, α1-6 or β1-3. These various subunits are binding sites for several drug classes that act as CNS depressants and for this reason are labeled as their receptors, eg, benzodiazepine (BZ) receptors. Unfortunately, nomenclature regarding this concept is confusing because references often identify several drug classes as agonists for the GABAA receptor, when more precisely they each bind to separate sites on the complex. For example, flumazenil is an antagonist only at benzodiazepine receptors and will not reverse the influences of GABA or drug classes that bind to alternative sites within the complex.
Benzodiazepines bind to several alpha subunits of the GABAA receptor complex.7,8 This does not result in opening of the chloride ion channel but potentiates opening in response to GABA. GABA triggers a burst of channel openings and these bursts increase in number if additional receptor sites are concurrently activated by benzodiazepines.7 It is significant that alone, benzodiazepines are incapable of opening chloride ion channels; they merely enhance the ionophore (channel) response to GABA. Other sedatives, such as propofol or barbiturates, are able to bind at other sites and open the channel independent of GABA. This difference offers one explanation for the relative safety of benzodiazepines. The intensity of their effect is ultimately determined by a restricted quantity of GABA, the brain’s normal endogenous neurotransmitter. These concepts are illustrated in Figure 2.
Researchers continue to gain a better understanding of the various subunits of the GABAA complex, and nomenclature defining the specific binding sites or receptors is evolving. Based on alpha1 and alpha2 subtypes, two benzodiazepine receptors have been suggested as BZ1 and BZ2. It is believed currently that BZ1 receptors mediate sedation and anticonvulsant effects, while anxiolysis, anterograde amnesia, and skeletal muscle relaxation are mediated by BZ2 receptors. As researchers continue to uncover additional receptor subtypes, there will be further clarification regarding distinct mechanisms that account for subtle differences in the range of effects provided by a particular benzodiazepine. Already the so-called non-benzodiazepines or ‘‘Z-compounds’’ such as zolpidem (Ambien) are claimed as more selective BZ1 agonists and are promoted for treatment of insomnia.
All benzodiazepines exhibit comparable efficacy as sedatives. In addition to their sedative effects, they also produce varying degrees of anterograde amnesia. Once recovered, patients have difficulty recalling events that occurred while they were sedated. It is significant that the patient, although sedated, was conscious when these events occurred. For example, a patient rendered unconscious by methohexital will not recall a palatal injection of lidocaine. This is not anterograde amnesia; he/she was unconscious. A patient sedated with a benzodiazepine might cry out and complain while receiving a palatal injection, but will often not recall the event when questioned postoperatively.
Benzodiazepines have little if any effect on respiration or cardiovascular function at doses recommended by manufacturers for use at home. This is consistent with doses used for minimal sedation. Although suicidal attempts with the benzodiazepines are relatively frequent, serious sequelae are rare unless other drugs are taken concomitantly.9,10 However, at doses used for moderate or deep sedation, this is no longer the case. All benzodiazepines will reduce ventilation in a dose-dependent manner, primarily by depressing hypoxemic drive. At high doses used to induce unconsciousness, transient periods of apnea are typical. Of greater significance is the fact that benzodiazepines decrease muscle tone in the upper airway, which fosters soft-tissue upper airway obstruction. They must be used cautiously in patients with chronic respiratory disorders such as chronic obstructive pulmonary disease or obstructive sleep apnea. Likewise, at greater doses, all benzodiazepines can lower blood pressure, which is generally accompanied by a reflex increase in heart rate. Caution is advised when sedating patients prone to hypotension and episodes of fainting.
Pharmacokinetic Considerations
Differences among benzodiazepines are largely pharmacokinetic and include their relative degree of lipid solubility and pattern of clearance, including elimination half-lives for parent drug and metabolites. These characteristics are the principal basis for selection because they influence the time of onset, duration of effect, and time for complete recovery following discharge.
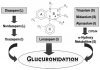
The most striking difference among benzodiazepines concerns their pattern of clearance. Some are biotransformed to active metabolites that have extended elimination half-lives, often longer than the parent drug. Metabolism of the benzodiazepines is the result of a variety of hepatic Phase 1 reactions: demethylation, hydroxylation, and oxidation. With the exceptions of lorazepam and oxazepam, benzodiazepines are metabolized by hepatic oxidative enzymes. The resulting metabolites, some of which retain anxiolytic activity, undergo glucuronide conjugation and elimination primarily through renal excretion. Oxazepam (Serax) and lorazepam (Ativan) are noteworthy in that their elimination pathway is via a one-step conjugation reaction, yielding water-soluble, pharmacologically inactive metabolites that are excreted in the urine. Their elimination is not affected significantly by altered liver function11 (Figure 3).
Unfortunately, some authors have spawned a misconception by citing elimination of half-lives as the basis for categorizing benzodiazepines as short, intermediate, and long-acting. While half-lives reflect the time required for drug elimination, they do not correlate well with the duration of sedation. Following single doses, such as those administered for procedural sedation, the onset and duration are related most to the drug’s lipid solubility. Those having greater lipid solubility have fast onset due to rapid absorption and diffusion through the blood-brain barrier. The duration of effect will be shorter also, because as the drug in serum distributes to adipose tissue, serum concentration declines and thereby promotes redistribution from the brain.1,12-14 This concept is evident when comparing diazepam and lorazepam in Table 4. Following repeated administration, elimination half-life may assume a more significant influence on the duration of clinical effect but, more importantly, influences the time required for complete recovery following discharge.
Clinical Considerations
When selecting a benzodiazepine for outpatient sedation, those that are highly lipid-soluble and have short elimination half-lives would appear to be more desirable than those eliminated more slowly. This is not always the case, however. While short-acting and rapidly cleared agents may be preferred for many patients, those patients suffering from chronic anxiety disorders may experience acute anxiety and increased postoperative sensitivity to pain if residual drug effects wear off too rapidly. For these individuals, agents having longer half-lives may be preferred. The decline in their serum concentration is more gradual, and this may provide a lingering anxiolytic effect throughout the ensuing day.
Diazepam is conventionally regarded as the prototypic benzodiazepine, but it is important to appreciate that all of these agents are comparable in providing sedation, provided equipotent dosages are used. Diazepam can be painful on intravenous (IV) injection. To reduce this discomfort and possible concern on the patient’s part, it should be administered into a rapid IV infusion and injected at a rate so slow that no discomfort is noted. It is also suggested that the largest vein accessible should be selected for venous cannulation. Veins on the dorsum of the hand should be avoided when possible. Diazepam undergoes hepatic biotransformation to active metabolites, nordiazepam, and oxazepam. These products may be concentrated in bile and secreted into the gut where they are reabsorbed.16 This is described as enterohepatic cycling and may result in a subsequent, though very much smaller peak effect several hours after a single IV dose of this drug.
Only two additional benzodiazepines, lorazepam and midazolam, are available for parenteral administration. Lorazepam has relatively low lipid solubility, which delays its onset to such an extent that titration to the sedative endpoint is difficult. Following IV injection, peak onset may require 20 to 30 minutes.16 Furthermore, it produces an extended duration of effect that may be troublesome in the ambulatory setting. For these reasons, diazepam and midazolam are the agents preferred for IV sedation. Significant data for commonly used benzodiazepines are presented in Table 4.
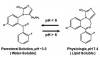
Midazolam has largely replaced diazepam as the benzodiazepine of choice for IV sedation. Compared to both diazepam and lorazepam, midazolam is not only more lipid soluble, but it has shorter distribution and elimination half-lives. These are similar for the parent compound and its only active metabolite, alpha hydroxymidazolam. When compared to diazepam, midazolam produces a more rapid onset, a slightly shorter duration, and perhaps a greater degree of amnesia.17 Midazolam is water soluble in solution and, unlike diazepam, is not dissolved in propylene glycol. For this reason, it is essentially painless during injection. Once injected and exposed to physiologic pH, however, its molecular structure assumes a highly lipid-soluble configuration.18 These molecular configurations are illustrated in Figure 4 and help to explain issues regarding the use of midazolam for oral sedation in pediatric patients. The acidity of gastric fluid favors the fraction of an oral dose that remains in the open-configured water-soluble state. This limits absorption and along with first-pass metabolism results in an oral bioavailability of approximately 40%. Midazolam is available as a commercially prepared syrup in a 2 mg/mL concentration and is administered most conventionally as a 0.5 mg/kg dose. However, Brosius and Bannister19 have shown superior bioavailability and sedation scores with an identical concentration of a self-prepared mixture using a standard parenteral midazolam formulation combined with a syrup (Syrpalta). Their mixture had a higher pH than the commercially prepared product, which likely fostered reconfiguration to a greater proportion of the closed-ring, lipid soluble midazolam molecules favoring absorption. This would likely be true as well using flavored acetaminophen syrup as the diluent. Simply withdraw 0.5 mg/kg midazolam from a 5 mg/mL concentration and mix with the manufacturer’s recommended dose for the flavored acetaminophen syrup. There is little advantage to using midazolam orally in adult patients unless they have difficulty with pill swallowing. Otherwise, the efficacy, onset, and duration for triazolam are virtually identical.
Following its introduction for general use, a significant number of mishaps were associated with the use of midazolam for sedation.20 Virtually all of these cases can be explained by failure to recognize its potency compared to diazepam and also its use in combination with opioids. The manufacturer further complicated matters by marketing the agent initially as a concentration identical to that of diazepam (5 mg/mL), rather than the 1 mg/mL concentration now recommended for intravenous use. Use of the 5 mg/mL concentration should be reserved for intramuscular (IM) and oral (PO) administration or for induction of unconsciousness in deep sedation techniques.
Estimates of midazolam potency vary from two to five times that of diazepam. White et al21 compared the potencies of diazepam and midazolam, and found that the dose-response for diazepam was much more gradual. As doses were increased, the difference in their potencies was greater. This finding provides one explanation for the disagreement regarding equipotent doses reported in the scientific literature. Furthermore, these results confirm present guidelines to carefully titrate midazolam in 1-mg increments. One must be particularly careful when administering any drug intravenously to the elderly or debilitated patient. Benzodiazepines in general and midazolam in particular are more likely to produce respiratory and cardiovascular depression in this population. In all probability, age-related reductions in hepatic blood flow and reduced enzyme activity make the elderly particularly sensitive to the effects of this drug.22 Increments should be halved when managing not only geriatric patients but those having significant medical compromise as well.
In recent years novel agents resembling benzodiazepines have been introduced for managing insomnia. These so-called ‘‘Z compounds’’ include zolpidem (Ambien), zaleplon (Sonata), and eszopiclone (Lunesta). Their molecular structures differ from benzodiazepines and can be distinguished chemically as nonbenzodiazepines for marketing a strategy to capitalize on negative views regarding benzodiazepines. They act as agonists at benzodiazepine receptors, exhibiting some preference for the BZ1 subtype, which implies they produce less amnesia, anxiolysis, or muscle relaxation. However, the clinician is wise to always view newly introduced products with some skepticism; the zeal of manufacturers for marketing often exceeds eventual scientific confirmation. While initially claimed to produce less risk for dependence, this has been contradicted by postmarketing data.7 Clinical considerations are identical to those discussed for benzodiazepines, and their pharmacokinetics closely resemble those for triazolam (Halcion). They have considerable lipid solubility that provides a rapid onset and short duration, short elimination half-lives (<4 hours), and no active metabolites.7,15 Zolpidem is available generically and 10 mg has been found equipotent to 0.25 mg triazolam.12
An advantage of benzodiazepines and the Z compounds over other classes of sedatives is that their effects can be reversed. Flumazenil (Romazicon) is a selective receptor antagonist that effectively reverses somnolence and respiratory depression attributed to benzodiazepines.15 It has no affinity for other receptors within the GABA receptor complex, and it will not reverse the actions of drug classes acting at these sites. Flumazenil is prepared as a 0.1-mg/mL concentration in either 5-mL or 10-mL vials. It can be administered intravenously in 0.2-mg increments every 3 to 5 minutes up to a total dosage of 1 mg. It can also be administered by sublingual injection, although its onset is slower.23 Duration of reversal ranges from 20 to 60 minutes, which could permit re-sedation if large doses of benzodiazepine have been administered. Re-sedation is most likely following large induction doses or cumulative doses during the course of a long procedure. It is highly unlikely following low or conventional doses of benzodiazepines, eg, less than 10 mg midazolam IV.16 To manage excessive somnolence or respiratory depression, attention should be given first to standard airway support including supplemental oxygenation and ventilation. Flumazenil is contraindicated for patients dependent on benzodiazepines and must be used cautiously for those having a history of chronic panic-anxiety disorders or convulsive seizures managed with benzodiazepines.
CHLORAL HYDRATE
Chloral hydrate is most often prescribed for oral sedation in children. Although somewhat active as the parent compound, chloral hydrate is rapidly converted to trichloroethanol, which is responsible for most of its effects. As can be deduced from its name, this sedative shares many characteristics with ethanol. Binding sites (receptors) for ethanol have been identified on the GABAA receptor complex, and chloral hydrate is presumed to promote GABA-mediated effects. Dosages required to produce effective sedation are frequently irritating to the gastrointestinal mucosa and nausea is not uncommon. Like ethanol, patients sedated with chloral hydrate may become delirious and combative when provoked by pain.7 This, unfortunately, has been the case when using this agent for managing highly recalcitrant pediatric dental patients. Although alternative sedatives are now available, chloral hydrate continues to be popular among pediatric dentists, a practice that originated prior to the introduction of benzodiazepines. At this time there is simply no logical basis to choose chloral hydrate over PO midazolam in pediatric patients.
ANTIHISTAMINES
Antihistamines are less effective than benzodiazepines as anxiolytics or sedatives, and for this reason they cannot be recommended as primary agents for procedural sedation. They have no actions related to the GABA receptor complex described for benzodiazepines. Their sedative effect is attributed to their action as antagonists at histaminergic and cholinergic receptor sites, countering the normal excitatory influences of these respective neurotransmitters within the CNS. This action can be a useful adjunct in potentiating sedation when benzodiazepines are ineffective alone. At conventional doses, respiratory depression and hemodynamic influences are negligible, but peripheral anticholinergic side effects and a central anticholinergic syndrome, including delirium, preclude the use of greater than conventional doses to achieve better sedation. A summary of antihistamines most commonly used in sedation regimens is presented in Table 5. Dosages greater than those provided offer no further advantage and increase risk for both delirium and central anticholinergic syndromes.
Histamine and acetylcholine are also among several neurotransmitters associated with neural pathways involved in nausea and vomiting. For this reason, antihistamines are useful antiemetic agents, but their use as prophylaxis for postoperative nausea and vomiting is questionable.24 Compared to other antihistamines, promethazine may be more effective in managing actual episodes of postoperative nausea and vomiting because it also blocks dopamine within the chemoreceptor trigger zone adjacent to the vomiting center. Emetic influences of opioids are attributed in part by activating these dopaminergic pathways. Unfortunately, dopamine antagonists also block these receptors within the basal ganglia and may produce extrapyramidal syndromes, a collective term for several conditions including Parkinsonism, tardive dyskinesia, and akathisia. Of these conditions, akathisia is the most common and presents as a subjective feeling of restlessness and a compelling need to move about. This behavior may be mistaken for agitation; distinguishing agitation from akathisia is critical to avoid an inclination to further sedate the patient. While extrapyramidal symptoms are bizarre, and generally frighten the patient and practitioner alike, they are never life-threatening. The added anticholinergic action of diphenhydramine is useful for countering acute episodes, should they occur.25 A final note on promethazine is worth mention. Like other phenothiazine derivatives, it has antagonist actions on vascular alpha receptors, which increases risk for postural hypotension, especially in the elderly.
Two central nervous system disorders should be considered when using antihistamines. The first of these is Parkinson disease, a degenerative disorder within the basal ganglia attributed to a relative dopamine deficiency and acetylcholine excess. The anticholinergic action of most antihistamines renders them attractive components of sedation regimens for patients having this disorder, provided they are not currently receiving other anticholinergic medication such as benztropine (Cogentin). Diphenhydramine has been suggested for this purpose.26 In contrast, promethazine should be avoided because it possesses modest but significant dopaminergic blocking activity. The second disorder for consideration is Alzheimer’s disease. This dementia is poorly understood but includes degeneration of cortical cholinergic neurons that function in cognition. It is wise to avoid all antihistamines or other agents having significant anticholinergic action for patients with Alzheimer’s disease or any other evidence of dementia.27
OPIOIDS
In addition to their analgesic effect, opioids mediate sedation by acting as agonists at both mu and kappa opioid receptors. This effect is less intense and certainly more unpredictable than that provided by sedative anxiolytics, but their combination may result in profound synergism. Vinik et al28 confirmed a dramatic, dose-dependent reduction in the hypnotic ED50 (effective dose for 50% of patients) when combining midazolam with alfentanil. Depression of respiratory centers parallels that in other brain regions, and these data provide additional insight into the added respiratory depression consistently observed when benzodiazepines and opioids are combined. For this reason the use of opioids cannot be recommended for PO sedation regimens and should only be used when carefully titrated during intravenous sedation techniques.
Opioids are commonly included in sedation regimens despite a conspicuous lack of scientific studies that confirm any advantage compared to using sedatives alone. In one of the few studies addressing this issue in moderate sedation, Dionne29 demonstrated a positive effect when managing difficult patients, but otherwise found that opioids provided little advantage over single, sedative-anxiolytic regimens. Many putative benefits derived from opioids in both moderate and deep sedation regimens are difficult to measure scientifically. Their analgesic effect is trivial in most cases because patients receive local anesthesia. Nevertheless, long and difficult dental procedures may result in annoying, noxious stimulation that may be attenuated by opioids, and sitting motionless for long periods with the mouth open wide can be equally uncomfortable. Further justification for the use of opioids in anesthesia and sedation is entrenched mostly on an empiric basis, predicated on several confirmed influences. The sedative effect of opioids is synergistic with that of most sedatives and analgesia may diminish the discomfort associated with local anesthetic injections. Opioids are also ‘‘cardioprotective’’ because they depress catecholamine release and obtund sympathetic reflexes to noxious stimuli. This influence is beneficial in particular for patients with hypertension, tachyarrhythmias, and ischemic heart disease. Considering the relative safety of carefully titrated doses, it is difficult to contradict their use for intravenous sedation regimens.
Meperidine and fentanyl are the opioids used most frequently, but the agonist-antagonist, nalbuphine, has gained considerable popularity. At conventional dosages, there is little, if any, difference in their sedative or analgesic effects. Unlike their analgesic effect, which follows a fairly consistent dose-response, sedative effects do not always follow this pattern, and high doses invariably lead to nausea and vomiting, especially during postoperative recovery and ambulation. For this reason, dosages in sedation regimens should be limited to those amounts presented, along with additional data, in Table 6.
Despite its historical popularity in sedation regimens, meperidine has several properties that render it less attractive than fentanyl or nalbuphine (Table 6). While its anticholinergic properties may provide a useful antisialagogue effect, it increases heart rate and also depresses myocardial contractility. Other opioids tend to reduce heart rate and have little or no influence on contractility.30 Its active metabolite, normeperidine, is a CNS stimulant having an elimination half-life of 16 to 18 hours compared to 3 to 8 hours for the parent drug. And finally, meperidine triggers greater release of histamine than most conventional opioids.31 Facial flushing or pruritus can be annoying for both patient and provider.
Fentanyl does not promote histamine release, which is a significant advantage over meperidine. It is highly lipid soluble and redistribution accounts for a relatively short duration of action (approximately 30 minutes) when administered in increments for intravenous sedation. Following repeated doses, fentanyl accumulates and its effects wane more in line with its elimination half-life, and this must be considered at discharge. Fentanyl has a greater potential than other opioids for producing skeletal muscle rigidity. This is most notable for muscles of the chest wall and the vocal cords. A patient experiencing chest or vocal cord rigidity may be unable to breathe, even when conscious, and cannot be ventilated manually. The incidence and severity of muscle rigidity is increased following rapid infusion of opioids and has been reported with as little as 50 mg of fentanyl when administered rapidly.32 The precise mechanism is unknown but, based on its reversal by naloxone, it is believed to be mediated by mu receptors at supraspinal sites.33 The appropriate use of naloxone is addressed below.
Nalbuphine does not produce euphoria and, in fact, may provoke dysphoria and delirium in some patients. Like other agonist-antagonists, nalbuphine demonstrates a ceiling dose-response that diminishes the risk for significant respiratory depression if high doses are used. This must be clarified, however. At conventional equipotent doses (nalbuphine 10 mg, meperidine 100 mg, and fentanyl 100 mg) all opioid effects are equivalent, including respiratory depression. As doses are increased, however, nalbuphine eventually achieves a ceiling effect, whereas the others will continue to produce a greater intensity of effects in a dose-response manner.30 Also in contrast to conventional mu agonists, nalbuphine has little potential for abuse, and it is not categorized by the drug enforcement agency (DEA) as a Class II narcotic. In fact, it is currently unscheduled and can be purchased without the inconveniences of DEA forms required when ordering meperidine or fentanyl. Because it lacks euphoric influences, it is ideal for patients having a prior history of drug abuse. However, it must be avoided if a patient has evidence of current opioid dependence from medical use or abuse because its action as an antagonist at mu receptors can precipitate symptoms of withdrawal. In the event this occurs, careful titration of a conventional mu agonist is required to relieve symptoms.
Naloxone is the prototypical, opioid receptor antagonist. It is capable of reversing the influences of both endogenous (eg, endorphins) and exogenous opioids. Following its administration, naloxone effectively reverses all opioid effects, including analgesia and suppression of catecholamine release. Abrupt reversal can trigger an acute sympathetic response including tachycardia, hypertension, and pulmonary edema.30 This concern is more significant during surgical procedures under general anesthesia than in the typical dental setting where adequate local anesthesia is present. For those practitioners without advanced training in managing unconscious patients, this issue should be disregarded. When confronted with an unconscious patient sedated with opioids, standard airway support should be provided and naloxone 0.4 mg administered as rapidly possible. For practitioners with advanced training, and provided the airway and ventilation are in control, it is reasonable to dilute the typical 0.4 mg/mL concentration in 10 mL and titrate in 0.04 mg to 0.08 mg (1 mL to 2 mL) increments until adequate ventilation is restored. Naloxone has a relatively short elimination half-life (approximately 1 hour), and the duration for reversal may be as brief as 30 minutes. If high accumulated doses of an opioid have been used, an IM injection of 0.4 mg may be considered for subsequent protection. However, this is needless when conventional opioid dosages have not been exceeded.
NITROUS OXIDE
General principles of nitrous oxide and other inhalation anesthetics have been reviewed more thoroughly in a previous continuing education article by this author and his colleague.34 Understanding of the mechanisms by which anesthetic gases produce general anesthesia is still evolving. Leading theories support multiple sites of action throughout the brain and spinal cord in producing each component of the general anesthetic state. In each case, ion fluxes are altered by influencing the activity of several neurotransmitters, primarily GABA, glycine, and glutamate.4,5 Unlike other anesthetics, nitrous oxide also produces a mild analgesic effect at subanesthetic concentrations. The mechanism for this effect most likely involves an interaction with the endogenous opioid system because it is abolished by administration of the opioid antagonist, naloxone. The strongest evidence is that nitrous oxide stimulates release of enkephalins, which bind to opioid receptors that trigger descending noradrenergic pathways.35
When using nitrous oxide it is important to appreciate the unit of potency for inhalation agents. This is expressed as minimum alveolar concentration (MAC) and represents the percent concentration required to produce immobility in response to a surgical stimulus for 50% of patients. It is analogous to the ED50 used for conventional drugs. Nitrous oxide has extremely low potency (MAC = 104), a concentration that is not achievable under normal conditions and fails to allow inclusion of ample oxygenation. This is in contrast to potent inhalation anesthetics such as desflurane and sevoflurane having MACs of 6 and 2, respectively. All inhalation anesthetics have influences on respiratory and cardiovascular function but, unlike the more potent agents, those attributable to nitrous oxide have little clinical significance at conventional concentrations.
Despite its lack of potency as a general anesthetic, sub-MAC concentrations of nitrous oxide (0.1 MAC to 0.5 MAC or approximately 10% to 50%) have an impressive record of safety when used to allay apprehension and anxiety during dental treatment. These concentrations are easily titrated and very safe. However, caution is advised because this contention does not address considerations when nitrous oxide is combined with other inhalation anesthetics, sedatives, or opioids. These agents not only lower the MAC for nitrous oxide, but work synergistically in depressing respiratory and cardiovascular function. For example, a 50-kg elderly patient premedicated with a sedative and opioid may require only 60% to 70% nitrous oxide to achieve MAC and respiratory depression may be more significant.
Titration of nitrous oxide is an ideal supplement to single-dose enteral regimens for procedural sedation. This strategy is far safer than attempting repeated oral or sublingual doses (stacking) of conventional sedatives. Titration of nitrous oxide can also be used safely along with multiple-drug intravenous techniques provided one respects the issues of synergism addressed above. It may also be attractive initially in highly anxious patients during venipuncture, and its ability to increase venous tone may facilitate difficult venous access.
Given its long history of safety, it could be argued that nitrous oxide is the safest of all the modalities available for sedation in dentistry. However, like any other pharmacologic agent, nitrous oxide may not be suitable for all patients. Clearly, an inability to tolerate a nasal mask poses an absolute contraindication. Generally, such patients fall into one of two categories—those who cannot inhale adequately through the nose because of anatomic or disease-induced nasopharyngeal obstructions, and those who resist placement of the nasal mask due to psychologic or cognitive disturbances. Nitrous oxide can increase pressure in closed air spaces and any compromise in patency of the Eustachian tube may lead to pressure increases within the middle ear.36 In fact, it has also been suggested that any recent surgery of the ear presents a contraindication for nitrous oxide.37
A final consideration is the use of nitrous oxide in pregnant patients. Clearly, all purely elective dental treatment should be avoided during pregnancy, especially during the first trimester. However, urgent dental care is frequently required for patients who are pregnant. Under these circumstances it is not unusual for the patient to be anxious and fearful, often extremely so. For these patients, apprehension should be allayed using the safest agents available, and nitrous oxide fulfills this requirement.38,39 For the pregnant patient who is apprehensive and requires urgent dental care, nitrous oxide should be regarded as the sedation agent of choice. Any evidence of complication during the pregnancy certainly warrants consultation with the patient’s obstetrician.
AGENTS LIMITED TO DEEP SEDATION TECHNIQUES
With the exception of volatile inhalation agents, there is no drug or class of drugs that can be considered a “general anesthetic,” nor is there one that can be labeled as specific for deep sedation. Drug dosage determines the level of sedation, and only those providers with advanced training should administer dosages that render a state of unconsciousness. Drugs discussed thus far have relatively shallow dose-response curves that enable their use for all intended levels of sedation. At low doses the following drugs can also produce minimal and moderate sedation, but their dose-response is steep and can easily produce unconsciousness as well. Furthermore, the barbiturates and propofol produce a far more significant depression of cardiovascular and respiratory function. Their use requires a designated provider to continuously assess respiration and cardiovascular status. The sum of these considerations has led most regulatory boards to restrict their use to providers having advanced formal training in deep sedation and general anesthesia. Table 7 provides a comparison of the relative respiratory and cardiovascular influences of drug classes discussed in this article.
METHOHEXITAL
Like benzodiazepines, barbiturates such as methohexital bind to receptors within chloride ion channels (Figure 2). At low doses they potentiate the action of GABA by prolonging the period of chloride channel opening. However, at higher doses they open chloride ion channels directly, independent of GABA. This additional action accounts for a lower therapeutic index compared to benzodiazepines.7,40 Using doses intended to produce moderate sedation, barbiturates are no more effective than benzodiazepines and lack significant anxiolytic and amnestic properties. Another drawback, especially at lower moderate sedation dosages, is that painful stimuli may incite delirium and agitation.7 However, when inducing hypnosis for deep sedation or general anesthesia induction, barbiturates are more predictable. Like thiopental, methohexital is very short-acting due to rapid redistribution, but it is associated with less postoperative sluggishness due to faster clearance. Compared to other barbiturates, methohexital is more likely to produce excitatory influences in the upper airway such as cough and hiccoughs.
PROPOFOL
Propofol is a non-barbiturate hypnotic that activates specific receptors within chloride ion channels normally regulated by GABA (Figure 2). Like barbiturates, it not only potentiates GABA but opens the chloride channels directly. Propofol is similar to methohexital in its rapid onset and duration but is cleared more rapidly due to both hepatic and extrahepatic sites of metabolism. For this reason, it produces a much faster recovery and is better suited for continuous infusion techniques due to its short context-sensitive half time.1 Unlike barbiturates, propofol is antiemetic and produces anterograde amnesia at sedative concentrations.40-42 The undiluted 1% formulation can be irritating to veins, but this can be countered or prevented by administering 1 mL (10 mg) of 1% lidocaine for intravenous use into the infusion line.
KETAMINE
Ketamine acts as an antagonist at a subtype of glutamate receptor called N-methyl-D-aspartate. These receptors are normally targeted by the excitatory neurotransmitter, glutamate, and provide for corticothalamic communication. Ketamine interrupts or disassociates this communication and is generally described as a dissociative general anesthetic agent when administered intravenously in 1-mg/kg to 2-mg/kg doses or in intramuscular doses of 5 mg/kg to 10 mg/kg.40,43 The anesthetic state produced is characterized by the production of unconsciousness, amnesia, analgesia, catalepsy, and, on occasion, vivid dreams. Because ketamine is a derivative of phencyclidine, a known hallucinogen, the occurrence of dreams in 15% to 30% of adults receiving the agent is not unexpected. These unpleasant effects can usually be prevented by including a benzodiazepine in the regimen.40,43-45
Ketamine is unique compared to other agents in several additional manners. Patients generally maintain protective airway reflexes, and it provides bronchodilation, making it useful in patients with asthma. However, it produces an excitatory influence on cardiovascular function, which contraindicates its use in patients with significant cardiac arrhythmias, coronary artery disease, or hypertension. Ketamine stimulates salivary and airway secretions, and for this reason an antisialagogue should be considered in the regimen. Glycopyrrolate 5 mg/kg up to 0.1 mg to 0.2 mg is a common choice.44,45
A technique using low doses of ketamine produces a state that has been termed by Bennett as dissociative sedation.46 IM injections of 1 mg/kg to 3 mg/kg produce a dissociative sedative state within 5 minutes that lasts about 15 to 20 minutes. This state is characterized by maintenance of consciousness accompanied by mental indifference, analgesia, and stable vital signs. Patients frequently behave in a ‘‘robotic’’ fashion. Requests to open the mouth, for example, are followed immediately in an exaggerated manner. Often the command followed by the patient will last for an extended period of time before a reminder is required. To assure complete analgesia, local anesthesia must be administered, but patients generally have no reaction to the injections. Green et al47 have also described safe and effective results using 4 mg/kg of ketamine by IM injection for pediatric patients managed in the emergency department. Nausea and vomiting are more common if IM doses are repeated.
REMIFENTANIL
The pharmacodynamics of remifentanil are virtually identical to those for fentanyl.48,49 Its advantage is its extremely rapid clearance by esterases within body tissues and erythrocytes. It is a poor substrate for plasma cholinesterases and poses no concern for patients with pseudocholinesterase deficiency. It may be described as the first “ultra-short acting” opioid and can be used only by continuous infusion techniques. Following discontinuation of an infusion, full emergence occurs within 5 to 15 minutes.48 When used in combination with propofol, as is commonly the case, infusion rates for both drugs should commence at the lowest ranges and titrated upward.
Dosages and serum concentrations for the drugs indicated in deep sedation techniques are provided in Table 8 and were derived from multiple sources.40,43,48 This information is intended only for those providers having advanced training. When using intermittent titration techniques for moderate to deep sedation, the loading dose range for moderate sedation is suggested.
About the Author
Dr. Becker is an Associate Director of Education, General Dental Practice Residency, at Miami Valley Hospital in Dayton, Ohio.
REFERENCES
1. Becker DE. Pharmacokinetics of sedation. Anesth Prog. 2011;58:166-172.
2. Carrasco G. Instruments for monitoring intensive care unit sedation. Crit Care. 2000;4:217-225.
3. Chernik DA, Gillings D, Laine H, et al. Validity and reliability of the Observer’s Assessment of Alertness/Sedation Scale: study with intravenous midazolam. J Clin Psychopharmacol. 1990;10:244-251.
4. Patel PM, Patel HH, Roth DM. General anesthetics and therapeutic gases. In: Brunton LL, Chabner BA, Knollmann BC (Eds.). Goodman and Gilman’s The Pharmacological Basis of Therapeutics. 12th ed. New York, NY: McGraw-Hill Companies Inc, 2011.
5. Perouansky M, Pearce RA, Hemmings HC Jr. Inhaled anesthetics: mechanisms of action. In: Miller RD, Eriksson LI, Fleisher LA, et al, associate eds. Miller’s Anesthesia. 7th ed. Philadelphia, PA: Elsevier, Churchill Livingstone, 2009.
6. American Society of Anesthesiologists Task Force on Sedation and Analgesia by Non-Anesthesiologists, Practice guidelines for sedation and analgesia by non-anesthesiologists. Anesthesiology. 2002;96:1004-1017.
7. Mihic SJ, Harris RA. Hypnotics and sedatives. In: Brunton LL, Chabner BA, Knollmann BC (Eds.). Goodman and Gilman’s The Pharmacological Basis of Therapeutics. 12th ed. New York, NY: McGraw-Hill Companies Inc, 2011.
8. Molinoff PB. Neurotransmission and the central nervous system. In: Brunton LL, Chabner BA, Knollmann BC (Eds.). Goodman and Gilman’s The Pharmacological Basis of Therapeutics. 12th ed. New York, NY: McGraw-Hill Companies Inc, 2011.
9. Jatlow P, Dobular K, Bailey D. Serum diazepam concentrations in overdose. Am J Clin Pathol. 1979;72:571-577.
10. Greenblatt DJ, Allen MD, Noel BJ, Shader RI. Acute overdosage with benzodiazepine derivatives. Clin Pharmacol Ther. 1977;4:497-514.
11. Greenblatt DJ. Clinical pharmacokinetics of oxazepam and lorazepam. Clin Pharmacokinet. 1981;6:89-105.
12. Greenblatt DJ, Harmatz JS, von Moltke LL, et al. Comparative kinetics and response to the benzodiazepine agonists triazolam and zolpidem: evaluation of sex-dependent differences. J Pharmacol Exp Ther. 2000;293:435-443.
13. Greenblatt DJ, Miller LG, Shader RI. Neurochemical and pharmacokinetic correlates of the clinical action of benzodiazepine hypnotic drugs. Am J Med. 1990;88:18s-24s.
14. Greenblatt DJ, Harmatz JS, von Moltke LL, et al. Age and gender effects on the pharmacokinetics and pharmacodynamics of triazolam, a cytochrome P450 3A substrate. Clin Pharmacol Ther. 2004;76:467-479.
15. Olin BR, Hebel SK, Dombek CE (eds.). Drug Facts and Comparisons 2011. St Louis, MO: Facts and Comparisons Inc. 2011.
16. Greenblatt DJ, Ehrenberg BL, Gunderman J, et al. Kinetic and dynamic study of intravenous lorazepam: comparison with intravenous diazepam. J Pharmacol Exp Ther. 1989;250:134-140.
17. Wright SW, Chudnofsky CR, Dronen SC, et al. Comparison of midazolam and diazepam for conscious sedation in the emergency department. Ann Emerg Med.1993;22:201-205.
18. Reves JG, Fragen RJ, Vinik HR, Greenblatt DJ. Midazolam: pharmacology and uses. Anesthesiology. 1985;62:310-324.
19. Brosius KK, Bannister CF. Midazolam premedication in children: a comparison of two oral dosage formulations on sedation score and plasma midazolam levels. Anesth Analg. 2003;96:392-395.
20. Bailey PL, Pace NL, Ashburn MA, et al. Frequent hypoxemia and apnea after sedation with midazolam and fentanyl. Anesthesiology. 1990;73:826-830.
21. White PF, Vasconez LO, Mathes SA, et al. Comparison of midazolam and diazepam for sedation during plastic surgery. Plast Reconstr Surg. 1988;81:703-712.
22. Greenblatt DJ, Abernethy DR, Locniskar A, et al. Effect of age, gender and obesity on midazolam kinetics. Anesthesiology. 1984;61:27-35.
23. Heniff MS, Moore GP, Trout A, et al. Comparison of routes of flumazenil administration to reverse midazolam-induced respiratory depression in a canine model. Acad Emerg Med. 1997;4:1115-1118.
24. Becker DE. Nausea, vomiting and hiccups: a review of mechanisms and treatment. Anesth Prog. 2010;57:150-157.
25. Vinson DR, Drotts DL. Diphenhydramine for the prevention of akathisia induced by prochlorperazine: a randomized, controlled trial. Ann Emerg Med. 2001;37:125-131.
26. Stone DJ, DiFazio CA. Sedation for patients with Parkinson’s disease undergoing ophthalmologic surgery. Anesthesiology. 1988;68:821.
27. Dierdorf SF, Walton JS. Anesthesia for patients with rare and co-existing diseases. In: Barash PG, Cullen BF, Stoelting RK (eds.). Clinical Anesthesia. 5th ed. Philadelphia, PA: JB Lippincott Co, 2006.
28. Vinik HR, Bradley EL, Kissin I. Triple anesthetic combination: propofol-midazolam-alfentanil. Anesth Analg. 1994;78:354-358.
29. Dionne RA. Differential pharmacology of drugs used for intravenous premedication. J Dent Res. 1984;63:842-847.
30. Yaksh TL, Wallace MS. Opioids, analgesia, and pain management. In: Brunton LL, Chabner BA, Knollmann BC (eds.). Goodman and Gilman’s The Pharmacological Basis of Therapeutics. 12th ed. New York, NY: McGraw-Hill Companies Inc, 2011.
31. Flacke JW, Flacke WE, Bloor BC, et al. Histamine release by four narcotics: a double-blind study in humans. Anesth Analg. 1987;66:723-730.
32. Vaughn RL, Bennett CR. Fentanyl chest wall rigidity syndrome—a case report. Anesth Prog. 1981;28:50-51.
33. Coda BA. Opioids. In: Barash PG, Cullen BF, Stoelting RK (eds.). Clinical Anesthesia. 5th ed. Philadelphia, PA: JB Lippincott Co, 2006.
34. Becker DE, Rosenberg M. Nitrous oxide and the inhalation anesthetics. Anesth Prog. 2008;55:124-131.
35. Zhang C, Davies MF, Guo TZ, Maze M. The analgesic action of nitrous oxide is dependent on the release of norepinephrine in the dorsal horn of the spinal cord. Anesthesiology. 1999;91:1401-1407.
36. Morgan GE, Mikhail MS, Murray MJ. Clinical Anesthesiology. 4th ed. New York, NY: Lange Medical Books/McGraw-Hill. 2006.
37. Munson ES. Complications of nitrous oxide anesthesia for ear surgery. Anesth Clin North Am. 1993;11:559-572.
38. Santos AC, Braveman FR, Finster M. Obstetric anesthesia. In: Barash PG, Cullen BF, Stoelting RK (eds.). Clinical Anesthesia. 5th ed. Philadelphia, PA: JB Lippincott Co, 2006.
39. Rosen MA. Management of anesthesia for the pregnant surgical patient. Anesthesiology. 1999;9:1159-1163.
40. Reves JG, Glass PSA, Lubarsky DA, et al. Intravenous anesthetics. In: Miller RD, Eriksson LI, Fleisher LA, et al, (associate eds.). Miller’s Anesthesia. 7th ed. Philadelphia, PA: Elsevier, Churchill Livingstone, 2009.
41. Shafer SL. Advances in propofol pharmacokinetics and pharmacodynamics. J Clin Anesth. 1993;5:14s-21s.
42. Veselis RA, Reinsel RA, Feshchenko VA, Wronski M. The comparative amnestic effects of midazolam, propofol, thiopental, and fentanyl at equisedative concentrations. Anesthesiology. 1997;87:749-764.
43. White PF, Romero G. Nonopioid intravenous anesthesia. In: Barash PG, Cullen BF, Stoelting RK (eds.). Clinical Anesthesia. 5th ed. Philadelphia, PA: JB Lippincott Co, 2006.
44. Reich DL, Silway G. Ketamine: an update on the first twenty-five years of clinical experience. Can J Anaesth. 1989;36:186-197.
45. White PF, Way WL, Trevor AJ. Ketamine: its pharmacology and therapeutic uses. Anesthesiology. 1982;56:119-136.
46. Bennett CR. Ketamine. In: Paris PM, Stewart RD (eds.). Pain Management in Emergency Medicine. East Norwalk, Conn: Appleton and Lange, 1988.
47. Green SM, Rothrock SG, Lynch EL, et al. Intramuscular ketamine for pediatric sedation in the emergency department: safety profile in 1,022 cases. Ann Emerg Med. 1998;31:688-697.
48. Fukuda K. Opioids. In: Miller RD, Eriksson LI, Fleisher LA, et al (associate eds.). Miller’s Anesthesia. 7th ed. Philadelphia, PA: Elsevier, Churchill Livingstone, 2009.
49. Egan TD, Lemmens HJ, Fiset P, et al. The pharmacokinetics of the new short-acting opioid remifentanil (GI87084B) in healthy adult male volunteers. Anesthesiology. 1993;79:881-892.