You must be signed in to read the rest of this article.
Registration on CDEWorld is free. Sign up today!
Forgot your password? Click Here!
Drug-induced gingival enlargement has been defined as an increase in gingival tissue size resulting in whole or in part from systemic drug use.1 Medications usually associated with this condition belong to three different therapeutic classes: calcium channel blockers, immunosuppressants, and anticonvulsants, although rare cases of gingival enlargement have been reported in association with valproic acid and erythromycin.2 In this article, special emphasis is given to calcium channel blockers, phenytoin, and cyclosporin, since these agents represent the earliest and most studied models of drug-induced gingival overgrowth.3
History
In 1939, Kimball recorded the first documented case of drug-induced gingival overgrowth.3 Phenytoin, an anticonvulsant, was introduced in 1938 for the treatment of epilepsy and, since that time, has been widely documented as a contributor to enlarged gingiva. This agent is considered the earliest and most investigated model used to understand the pathophysiology of drug-induced gingival overgrowth.3
Cyclosporin-A (CsA) is a fungal metabolite produced by the fermentation of the species Trichoderma polysporum and Clyndocarpon lucidum.4,5 It was discovered by Borel et al in 1977, and is used to prolong the survival of allogenic transplants in humans. CsA reversibly inhibits immunocompetent lymphocytes, such as T-lymphocytes, without affecting the humoral immune response.6 In addition to gingival enlargement, side effects can include cardiotoxicity and nephrotoxicity. In 1939, Kimball first reported gingival overgrowth as a result of cyclosporine intake in renal transplant patients.3
Calcium channel blockers have been used since 1978 to treat symptoms of angina pectoris, ventricular arrhythmias, and post-myocardial syndrome. The first report linking calcium channel blockers to gingival overgrowth was published in 1984 by Ramon et al, who observed a population of hypertensive patients taking nifedipine for prolonged periods of time.2
Epidemiology
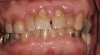
Drug-induced gingival enlargement is typically described as nodular, firm in consistency, and hard upon palpation. Clinical manifestations can be more severe in areas where local irritants such as plaque, defective restorations, and orthodontic or prosthetic appliances are present (Figure 1).7 Drug-induced gingival overgrowth is quite different between animals and humans. As demonstrated in rat models, the incidence of macroscopic gingival changes is usually close to 100% in all three of the previously mentioned classes of medication.8 In humans, however, the incidence has been reported to vary according to medication: 10% to 50% for phenytoin, 8% to 70% for cyclosporin-A, and 0.5% to 83% for nifedipine.6
Within each class of drugs, different agents may exhibit varying amounts of gingival enlargement. Among the calcium-channel blocking agents, for example, nifedipine typically exhibits the greatest amount of enlargement,9 followed by diltiazam9, amlodipine,10 and isradipine.11 Drug combinations can lead to an increased incidence of gingival overgrowth, as demonstrated in patients taking both cyclosporin and calcium channel antagonists, where gingival enlargement was present in more than 60% of patients.12 O’Valle et al reported a two-fold incidence of gingival modifications in patients taking CsA and nifedipine, compared to patients taking CsA alone.13
The incidence of drug-induced gingival overgrowth is higher in adolescents and young adults and usually becomes clinically evident in less than 3 months from the beginning of therapy.14 However, those findings could not be confirmed by an in-vitro study where gingival fibroblast responsiveness to phenytoin and cyclosporin was found not to be age-dependent. A sex-related responsiveness is evident in humans and in animals as well. Barak et al reported that the male-to-female ratio of a responder group of patients treated with nifedipine was 5:1.15 Although that was a small clinical study, those findings were consistent with results from other investigations where CsA- and phenytoin-induced gingival overgrowth were seen more frequently in younger males.16
Etiology/Pathophysiology
Histometric analysis of drug-induced gingival lesions reveals an increase in the number of cells as well as an increase in extracellular matrix, in contrast to a net increase in cellular size or number (hypertrophy and hyperplasia, respectively). The term overgrowth is preferred for histological characterization of such drug-induced gingival conditions.17
Despite many hypotheses, the biologic mechanisms responsible for drug-mediated gingival enlargement remain unclear. However, the available evidence supports a multifactorial etiology.18 In a review article, Brown et al hypothesized that three entities are necessary, but not sufficient, for drug-induced enlargement: the drug; bacterial-induced inflammation; and teeth.19 While it is clear that many factors may contribute to the inter-patient variability observed in gingival enlargement—including age, plaque, genetic predisposition, and local tissue properties—there is general agreement that tissue changes occur due to an interaction between the drug and the resident gingival fibroblasts.20
The Role of Plaque in Drug-Induced Gingival Overgrowth
Gingival enlargement is clinically similar regardless of the associated agent, but some inter-patient variability may be affected by the patient’s ability to adequately perform plaque control measures.21 Although controversy exists concerning the contribution of oral hygiene to the etiology of drug-induced gingival overgrowth, there is abundant clinical evidence of partial-to-complete clinical resolution of enlargement when proper plaque control is performed.22 Ellis et al, using regression analysis, found a significant relationship between gingival index and overgrowth severity, where reduction in the gingival index was followed by a corresponding decrease in the severity of gingival overgrowth.23 Those findings were confirmed by an in-vitro study where it was demonstrated that collagen synthesis was significantly increased in fibroblasts cultured with both nifedipine (10-7 mol/l) and high concentrations of IL-1β (50 and 500 pg/ml), compared to fibroblasts cultured with only nifedipine.24 Exposure of cell cultures to lipopolysaccharides from periodontopathic bacteria has been reported to enhance the release of pro-inflammatory cytokines, especially IL-1β.25 Consequently, this may partially explain the relationship between enlargement and plaque accumulation. Despite in-vitro studies corroborating that relationship, the extent to which plaque may be an etiologic factor remains uncertain, since plaque control and the removal of local irritation fails to consistently prevent the occurrence of gingival overgrowth.26
An investigation by Nakou et al that analyzed subgingival microflora associated with gingival enlargement found significant differences in the types of bacterial species that were evident in the gingival enlargement group when compared to the control group of patients taking nifedipine but experiencing no enlargement.27 Eubacteriace, Campylobacter concisus, Capnocytophaga spp. (Ochracea, sputigena, and gingivalis), F nucleatum, and F varium were among the species that were found most frequently in the responder group. The associated microflora in gingival sulci when overgrowth was present consisted mainly of Gram-negative pathogenic bacteria, indicating similarities with the microflora of destructive periodontal lesions.28
Such theories remain speculative, since it has not been possible to demonstrate whether changes in microflora are the cause or the effect of the enlargement. As noted by Modéer et al29 and Nery et al,30 plaque would act as a co-factor, since drug-induced gingival overgrowth is also observed in patients with excellent oral hygiene. Consequently, the authors concluded that rigorous plaque control in patients treated with certain drugs can limit the severity of lesion development but cannot prevent it altogether (Figure 2).
Pharmacokinetic Variables
Variations in drug kinetics also may be responsible for the wide clinical range in severity of gingival overgrowth observed in humans, as well as for the differences in the drug concentration of blood and crevicular fluid among individuals.31 In a population of institutionalized epileptic and post-transplant patients, stimulated saliva was collected to test phenytoin and cyclosporine concentration. Gingival crevicular fluid (GCF) is another important factor in the mechanism of gingival overgrowth. Ellis et al were first to demonstrate nifedipine and amlodipine sequestration within the overgrowth of gingival tissues.32 The pharmacokinetics of patients who had received nifedipine for at least 6 months were studied, and the incidences of gingival changes were found to be approximately 60%, with no significant difference between responders and non-responders. In responder patients, nifedipine concentration ranged between 15 and 316 (mean 84) times higher in GCF than in plasma, while non-responder patients had undetectable GCF nifedipine levels. Capillary gas chromatography confirmed the level of nifedipine concentration in the GCF. Nifedipine concentration in the GCF varied from 920 µg/l to 9300 µg/l, which represented a 15- to 316-fold increase from the plasma drug concentration.33 Ellis et al concluded that, under some circumstances, gingival tissue tends to concentrate nifedipine. Very high levels of nifedipine within the gingival tissue may predispose gingiva to nifedipine toxicity.32
Alterations in Connective Tissue Homeostasis
An essential feature of drug-induced gingival enlargement is an increase in the connective tissue matrix, likely associated with some form of alteration in connective tissue homeostasis. In-vitro studies have shown that drug-related alteration of collagen gene expression may be an important factor in the pathogenesis of gingival overgrowth; however, no clear pattern emerges. Both nifedipine and phenytoin could significantly induce the expression of collagen type I and type IV.34 Cyclosporine can increase collagen synthesis in cultured fibroblasts as well.35
Enhanced protein synthesis and the production of an inactive collagenase were observed in subpopulations of cultured fibroblasts. Shikata et al found an increased expression of type VI collagen genes when specific fibroblastic cell lines were cultured in the presence of phenytoin and nifedipine.36 While immunolocalization studies demonstrated abnormal accumulation of type VI collagen around collagen fiber bundles, the total RNA extracted from fibroblasts and tissues of enlarged gingiva demonstrated an increased type VI collagen RNA level, supporting the hypothesis of overactive collagen synthesis in those fibroblasts. The existence of subpopulations of fibroblasts that preferentially proliferate in response to phenytoin also has been hypothesized by Hassel et al in two in-vitro studies.37,38
An in-vitro study by Gultchin and Soshkan demonstrated that phenytoin-induced gingival overgrowth is related to a decrease in collagen breakdown, as opposed to an increase in synthesis.39 Kataoka et al, using flow cytometry with murine gingival fibroblasts, also described a reduction in collagen phagocytosis by gingival fibroblasts cultured with phenytoin and nifedipine. Thus, collagenolytic enzymes could play a key role in the homeostasis of gingival tissue metabolism, and its dysfunction could be a likely pathogenic factor involved in drug-induced gingival overgrowth.40 However, in-vitro studies have failed to reach a consensus about the effect of gingival enlargement-associated drugs on fibroblast collagenase. Collagenase activity measured in terms of C14 collagen degradation was found to be increased in fibroblasts cultured with nifedipine, decreased in fibroblasts cultured with phenytoin, and unchanged in fibroblasts cultured with cyclosporin.24
Although the three classes of drugs associated with gingival overgrowth are unrelated to each other, they are all known to affect intracellular calcium levels by inhibiting its entry into the cell.41 A certain intracellular concentration of calcium is required by fibroblasts for the synthesis and secretion of enzymes involved in collagen degradation, such as matrix metalloproteinases (MMPs). The upregulation of MMP gene expression is specifically triggered by protein kinase C via induction of the binding of the transcription factor AP-1 to a specific promoter sequence of the MMPs gene.42 Activation of protein kinase C is a calcium-dependent process and may explain, at the transcriptional level, how each of the gingival overgrowth-associated drugs may alter the sequence of events that lead to MMP expression and its release from the gingival fibroblasts.43 The diminished collagen breakdown would result in a shift of homeostasis, and a net accumulation of extracellular matrix would become clinically evident as gingival overgrowth. This hypothesis has been reinforced by reduced MMP-1 staining in drug-induced gingival overgrowth tissue samples.44
CsA does not increase collagen expression and seems unable to alter collagenase levels.45 Instead, the net increase in collagen associated with CsA is apparently due to reduced collagen degradation by the fibroblasts, as confirmed by several in-vitro and in-vivo studies.12,46 Collagen breakdown by fibroblasts occurs by two mechanisms: the phagocytic pathway and the MMP-dependent pathway.47 Cyclosporin-A may alter both pathways, reducing phagocytosis or inhibiting the secretion of MMPs by gingival fibroblasts.
Efficient phagocytosis is dependent upon calcium release from intracellular stores such as rough endoplasmic reticulum and mitochondria.12 Previous studies have demonstrated the ability of CsA to interfere with intracellular Ca2+ signaling of myocardic cells. The same mechanism is thought to play a role in CsA-induced cardiotoxicity.12 The initial steps of collagen phagocytosis rely on adhesive interactions between fibroblasts and collagen. As surface receptors, α-integrins play a key role in this interaction, and their affinity for collagen is regulated by intracellular calcium levels. This regulation takes place by means of a calcium-dependent feedback loop that may alter the integrin’s affinity for collagen and other matrix molecules.12,48 Cyclosporin-A may inhibit collagen phagocytosis by affecting mitochondrial and rough endoplasmic reticulum (RER), which stores calcium and ultimately controls intracellular homeostasis. Arora et al used an in-vitro model to analyze the effect of CsA on collagen bead phagocytosis with human and rat gingival fibroblasts and determined that CsA caused a dose-dependent inhibition of phagocytosis.48 Chelation of intracellular calcium with a common chelant, such as BAPTA/AM, also reduced collagen bead phagocytosis. Direct measurement of calcium levels in RER revealed that CsA completely blocked calcium discharge from RER stores, substantiating the hypothesis that CsA inhibits phagocytosis by deregulating intracellular calcium signaling. The CsA-induced attenuation of this calcium signal may be sufficient to inhibit collagen binding to collagen receptors, perhaps by regulating the affinity of the α2β1 integrin.49
Other in-vitro studies have been directed to the gingival non-collagenous matrix. A three-fold increase in the dry weight of non-collagenous matrix has been observed in gingival tissue treated with phenytoin.50 Increases in hexosamine, uronic acid and total protein per total gingival net weight—as well as increases in glycosaminoglycan and proteoglycan concentration—have been described in gingival samples from patients taking phenytoin.51
It is unknown why other connective tissues fail to grow in a manner similar to gingival tissue. Two possibilities theorized by Henderson et al may explain a site-specific alteration of human gingival fibroblast metabolism.52 The first hypothesis is that a particular responder fibroblastic phenotype localized in gingival tissue might exist. Only the responder fibroblasts residing in this tissue would be affected by the medications, whereas fibroblasts originating from other tissues (dermis, capsular ligament, etc.) would not be altered. The second hypothesis theorized that a different basal rate of collagen production exists among different populations of fibroblasts, regardless of the presence of a specific medication. In this situation, a generalized drug-induced overexpression of collagen would be clinically evident only in those populations of fibroblasts whose basal collagen synthetic rate was already elevated due to high tissue turnover (ie, gingival tissue fibroblasts).
Role of Growth Factors and Hormones
The role of growth factors has been elucidated in the pioneering study by Modéer et al. Gingival fibroblasts sampled from patients who had been administered phenytoin were shown to display an increase in cell-surface epithelial growth factor (EGF) receptors. Alterations in connective tissue homeostasis would be mediated by EGF through upregulation of its cellular receptor.53 Epidermal growth factor is a polypeptide synthesized by a wide range of normal cell types and exerts many biological effects. It is mainly secreted by the salivary and mammary glands and stimulates the proliferation and differentiation of epithelium- and mesenchyme-derived tissues, as well as protein and hyaluronic acid synthesis. Furthermore, EGF stimulates proliferation and collagen synthesis in cultured fibroblasts.54
These distinctive properties have led to the idea that EGF may play an important role in tissue remodeling and is likely responsible for increased fibroblast proliferation and collagen production in enlarged gingival tissue.55 To exert its effect, EGF must bind to a specific cell surface receptor, a single polypeptide chain that spans the plasma membrane of fibroblasts.56 Buduneli et al proposed an in-vitro model by which CsA upregulates the expression of EGF receptors on gingival fibroblasts, amplifying the cellular response to the growth factor. This mechanism would ultimately lead to increased proliferation and collagen synthesis and very well may be a component of the gingival tissue alterations observed in CsA-induced gingival enlargement.57 A similar upregulation of EGF-receptors was observed by Modéer et al in fibroblasts cultured from phenytoin-responder patients. The authors showed a 30% increase in the number of receptors in responders, whereas in non-responders a decrease in receptor number was observed.58
The role of EGF in CsA-associated overgrowth has been further investigated by Markopoulos et al. Seventeen patients undergoing cyclosporine treatment with clinically significant gingival overgrowth were tested and compared to an age- and sex-matched control group of patients taking CsA but without gingival alterations. Unstimulated whole saliva and GCF were collected from all individuals to assess EGF concentration. The growth factor concentration was found to be significantly higher in whole saliva of patients with cyclosporine-induced gingival enlargement compared to the control group. EGF was not detected in samples of GCF in either group. The fact that EGF was detected in saliva and not in GCF further supported the origin of EGF in the oral cavity from the salivary gland parenchyma.54
An upregulation in platelet-derived growth factor (PDGF) secretion was demonstrated when rat peritoneal macrophages and lymphocytes were cultured with phenytoin in a study by Dill et al, which showed that incubation of rat and human monocytes with phenytoin caused a significant elevation in the secretion of PDGF.59 This polypeptide is a major mitogen and chemoattractant for fibroblasts, inducing fibroblast proliferation and synthesis of GAGs, proteoglycans, fibronectin, and collagen.60 PDGF is secreted by macrophages and also released from platelets, endothelial cells, smooth muscle cells, and fibroblasts. It also may constitute a putative mechanism in gingival tissue enlargement.61
Transforming growth factor β is a multifunctional peptide that regulates diverse biologic activities including cell growth, apoptosis, cell differentiation, and extracellular matrix synthesis. TGF-β1 seems to be a key mediator of tissue fibrosis related to extracellular matrix accumulation in conditions such as gingival fibromatosis and in progressive renal diseases such as CsA-induced nephropathy.47 TGF-β1 also induces the release of calcium from endoplasmic reticulum and mitochondria. Consequently, the decrease in calcium-mediated phagocytosis under CsA treatment may be explained by the TGF-β over-expression and the secondary depletion of intracellular calcium storage.12,47 Cotrim et al investigated the effect of CsA on TGF-β, matrix metalloproteinases (MMPs) and their tissue inhibitor (TIMPs) production. The results showed that the addition of CsA to the culture media, at the same hematic concentration found in patients undergoing CsA treatment, stimulated TGF expression and decreased the expression of MMPs (collagenases). The effect on TIMPs was not significant. The author postulated that TGB-β modulates fibroblasts in an autocrine fashion.47 Despite those results, the exact role played by local growth factors is mostly unclear.
Histologic Characteristics
Both the epithelium and the connective tissue seem to be affected by the drugs. The lamina propria typically displays an increased vascularity and a chronic inflammatory cell infiltrate. Bundles of collagen fibers are locally densely packed or loosely textured and apparently immature. The epithelium is thickened and acantotic.45
Rateitschick-Pluss et al biopsied CsA-associated gingival lesions in humans and observed an abundant connective tissue covered with an irregular, multilayered, parakeratinized epithelium of varying thickness. The authors described penetration of epithelial ridges deep into a highly vascular connective tissue. Irregularly arranged collagen fibers also were observed along with focal infiltration of inflammatory cells.62 Lucas et al documented an ultrastructural study of nifedipine-induced enlarged gingiva.63 The tissue was characterized by a sparse perivascular inflammatory infiltrate and a diffuse mixture of dense collagen bundles interspersed with varying amounts of ground substance. Abundant stellate-shaped fibroblasts rich in secretory granules were described as well. Within the cytoplasm of these fibroblasts was a well-developed rough endoplasmic reticulum and a moderate number of mitochondria, suggesting an active state of collagen synthesis.63 Ayanoglou et al reported a histological, ultrastructural, and histomorphometric evaluation of nifedipine- and cyclosporin-induced gingival overgrowth.46 Six Sprague Dawley rats were administered both nifedipine and cyclosporin for 19 weeks. The histological and ultrastructural evaluation revealed the presence of numerous irregularly shaped blood vessels. Collagen bundles were abundant and parallel to the long axis of the tooth. Inflammatory cells such as macrophages and mast cells were observed adjacent to the junctional epithelium. Focal areas of hyalinization were observed within the connective tissue, and multinucleated cells were reported as well.
Conclusion and Clinical Considerations
The pathogenesis of drug-induced gingival overgrowth is multifactorial, and clinicians should be aware that patients may be taking medications that might result in gingival enlargement. Potential contributing factors include age, plaque control, pharmacokinetic variables, dosage, and duration of drug intake. A lack of standardized in-vitro investigations or appropriate animal models makes conclusions about the precise etiology of gingival overgrowth difficult.
Treatment alternatives include periodic periodontal maintenance if enlargement is minimal, esthetics is not a concern, probing depths are normal, bleeding upon probing is absent, and oral hygiene is satisfactory. In other cases, nonsurgical and surgical periodontal therapy can be considered. Restorative dentistry also can be performed to correct deficient restorations, improve contours, and facilitate effective plaque control. In addition, treatment of gingival enlargement may be influenced by the patient’s expectations, as well as medical and dental treatment history. Consultation with the patient’s physician may be appropriate to determine if changing medications (within a class or to another class of drugs) might be feasible. For example, carbamazepine for phenytoin; tacrolimus for CsA; and different calcium channel blocking agents—or antihypertensive agents—for nifedipine. In any event, further studies are indicated to more fully elucidate the pathophysiology of drug-induced gingival enlargement.
ABOUT THE AUTHORS
Michelle L. Moffitt, RDH
Adjunct Instructor, Department of Periodontics and Endodontics, University at Buffalo, The State University of New York, School of Dental Medicine, Buffalo, New York
Davide Bencivenni, DDS, MS
School of Dental Medicine, University of Modena and Reggio Emilia, Modena, Italy
Robert E. Cohen, DDS, PhD
Professor, Department of Periodontics and Endodontics, University at Buffalo, The State University of New York, School of Dental Medicine, Buffalo, New York
REFERENCES
1. American Academy of Periodontology. Glossary of Periodontal Terms. 4th ed. Chicago, IL: American Academy of Periodontology; 2001.
2. Ramon Y, Behar S, Kishon Y, Engelberg IS. Gingival hyperplasia caused by nifedipine—a preliminary report. Int J Cardiol. 1984;5(2):195-206.
3. Kimball OP. The treatment of epilepsy with sodium diphenyl hydantoinate. JAMA. 1939;112:1244-1245.
4. Borel JF, Feurer C, Magnée C, Stähelin H. Effects of the new anti-lymphocyte peptide cyclosporin A in animal . Immunology. 1977;32(6):1017-1025.
5. Colombo D, Ammirati E. Cyclosporine in transplantation - a history of converging timelines. J Biol Regul Homeost Agents. 2011;25(4):493-504.
6. Seymour RA, Smith DJ, Rogers SR. The comparative effects of azathioprine and cyclosporine on some gingival health parameters of renal transplant patients: A longitudinal study . J Clin Periodontol. 1987;14(10):610-613.
7. Lederman D, Lumerman H, Reuben S, Freedman PD. Gingival hyperplasia associated with nifedipine therapy. Report of a case. Oral Surg Oral Med Oral Pathol. 1984;57(6):620-622.
8. Ishida H, Kondoh T, Kataoka M, et al. Factors influencing nifedipine-induced gingival overgrowth in rats. J Periodontol. 1995;66(5):345-350.
9. Fattore L, Stablein M, Bredfeldt G, et al. Gingival hyperplasia: a side effect of nifedipine and diltiazem. Spec Care Dentist. 1991;11(3):107-109.
10. Triveni MG, Rudrakshi C, Mehta DS. Amlodipine-induced gingival overgrowth. J Indian Soc Periodontol. 2009;13(3):160-163.
11. Westbrook P, Bednarczyk EM, Carlson M, et al. Regression of nifedipine-induced gingival hyperplasia following switch to a same class calcium channel blocker, isradipine. J Periodontol. 1997;68(7):645-650.
12. Thompson JM, Seymour RA, Ellis JS, et al. Iatrogenic gingival overgrowth in cardiac transplantation. J Periodontol. 1995;66(8):742-746.
13. O’Valle F, Mesa F, Aneiros J, et al. Gingival overgrowth induced by nifedipine and cyclosporin A. Clinical and morphometric study with image analysis. J Clin Periodontol. 1995;22(8):591-597.
14. Seymour RA, Jacobs DJ. Cyclosporin and the gingival tissues. J Clin Periodontol. 1992;19(1):1-11.
15. Barak S, Engelberg IS, Hiss Z. Gingival hyperplasia caused by nifedipine: histopathological findings. J Periodontol. 1987;58:639-642.
16. Nishikawa S, Nagata T, Morisaki I, et al. Pathogenesis of drug-induced gingival overgrowth. A review of studies in the rat model. J Periodontol. 1996;67(5):463-471.
17. Hassell TM, Page RC, Lindhe J. Histological evidence for impaired growth control in diphenylhydantoin gingival overgrowth in man. Arch Oral Biol. 1978;23(5):381-384.
18. Seymour RA, Thomason JM, Ellis JS. The pathogenesis of drug-induced gingival overgrowth. J Clin Periodontol. 1996;23(3 Pt 1):165-175.
19. Brown RS, Beaver WT, Bottomley WK. On the mechanism of drug-induced gingival hyperplasia. J Oral Pathol Med. 1991;20(5):201-209.
20. Morisaki I, Mihara DJ, Kato K, et al. Phenytoin-induced gingival overgrowth in rats infected with Streptococcus sobrinus 6715. Arch Oral Biol. 1990;35(9):753-758.
21. Angelopoulos AP, Goaz PW. Incidence of diphenylhydantoin gingival hyperplasia. Oral Surg Oral Med Oral Pathol. 1972;34(6):898-906.
22. Barclay S, Thomason JM, Idle JR, Seymour RA. The incidence and severity of nifedipine-induced gingival overgrowth. J Clin Periodontol. 1992;19(5):311-314.
23. Ellis JS, Seymour RA, Steele JG, et al. Prevalence of gingival overgrowth induced by calcium channel blockers: a community based study. J Periodontol. 1999;70(1):63-67.
24. Johnson RB, Zebrowski EJ, Dai X. Synergistic enhancement of collagenous protein synthesis by human gingival fibroblasts exposed to nifedipine and interleukin-1-beta in vitro. J Oral Pathol Med. 2000;29(1):8-12.
25. Yoshimura A, Hara Y, Kanedo T, Kato I. Secretion of IL-1 beta, TNF-alfa, IL-8 and IL-1ra by human polymorphonuclear leukocytes in response to lipopolysaccharides from periodontopathic bacteria. J Periodontal Res. 1997;32(3):279-286.
26. Seymour RA, Smith DG. The effect of a plaque control programme on the incidence and severity of cyclosporine-induced gingival changes. J Clin Periodontol. 1991;18(2):107-110.
27. Nakou M, Kamma JJ, Andronikaki A, Mitsis F. Subgingival microflora associated with nifedipine-induced gingival overgrowth. J Periodontol. 1998;69(6):664-669.
28. Holt SC, Bramanti TE. Factors in virulence expression and their role in periodontal disease pathogenesis. Crit Rev Oral Biol Med. 1991;2(2):177-281.
29. Modéer T, Dahllöf G. Development of phenytoin-induced gingival overgrowth in non-institutionalized epileptic children subjected to different plaque control programs. Acta Odontol Scand. 1987;45(2):81-85.
30. Nery EB, Edson RG, Lee KK, et al. Prevalence of nifedipine-induced gingival hyperplasia. J Periodontol. 1995;66(7):572-578.
31. Daley TD, Wysocki GP, Day C. Clinical and pharmacologic correlations in cyclosporine-induced gingival hyperplasia. Oral Surg Oral Med Oral Pathol. 1986;62(4):417-421.
32. Ellis JS, Seymour RA, Monkman SC, Idle JR. Gingival sequestration of nifedipine in nifedipine-induced gingival overgrowth. Lancet. 1992;339(8806):1382-1383.
33. Ellis JS, Monkman SC, Seymour RA, Idle JR. Determination of nifedipine in gingival crevicular fluid: a capillary gas chromatographic method for nifedipine in microlitre volumes of biological fluid. J Chromatogr. 1993;621(1):95-101.
34. Romanos GE, Strub JR, Bernimoulin JP. Immunohistochemical distribution of extracellular matrix proteins as a diagnostic parameter in healthy and diseased gingiva. J Periodontol. 1993;64(2):110-119.
35. Ellis JS, Seymour RA, Thomason JM, et al. Periodontal variables affecting nifedipine sequestration in gingival crevicular fluid. J Periodontal Res. 1995;30(4):272-276.
36. Shikata H, Utsumi N, Shimojima T, et al. Increased expression of type VI collagen genes in drug-induced gingival enlargement. FEBS Lett. 1993;334(1):65-68.
37. Hassel TM. Evidence for production of an inactive collagenase by fibroblasts from phenytoin-enlarged human gingiva. J Oral Pathol. 1982;11(4):310-317.
38. Hassell TM, Provenza DV, Foster RA. Synthetic activities of mass cultures and clones of human gingival fibroblasts. Experientia. 1986;42(1):66-69.
39. Goultchin J, Shoskan S. Inhibition of collagen breakdown by diphenylhydantoin. Biochim Biophys Acta. 1980;631(1):188-191.
40. Kataoka M, Shimizu Y, Kunikiyo K, et al. Nifedipine induces gingival overgrowth in rats through a reduction in collagen phagocytosis by gingival fibroblasts. J Periodontol. 2001;72(8):1078-1083.
41. Gelfand EW, Cheung RK, Grinstein S, Mills GB. Characterization of the role for calcium influx in mitogen-induced triggering of human T-cells. Identification of calcium-dependent and calcium-independent signals. Eur J Immunol. 1986;16(8):907-912.
42. Birkedal-Hansen H. Role of cytokines and inflammatory mediators in tissue destruction. J Periodontal Res. 1993;28(6 Pt 2):500-510.
43. Hardie DG. Biochemical Messengers: Hormones, Neurotransmitters and Growth Factors. London, UK: Chapman & Hall; 1990:191-247.
44. Thomason JM, Sloan P, Seymour RA. Immunolocalization of collagenase (MMP-1) and stromelysisn (MMP-3) in the gingival tissue of organ transplant patients medicated with cyclosporin. J Clin Periodontol. 1998;25(7):554-560.
45. McGaw WT, Porter H. Cyclosporine-induced gingival overgrowth: an ultrastructural stereologic study. Oral Surg Oral Med Oral Pathol. 1988;65(2):186-190.
46. Ayanoglou CM, Letsy C. Cyclosporin A-induced gingival overgrowth in the rat: a histological, ultrastructural and histomorphometric evaluation. J Periodontal Res. 1999;34(1):7-15.
47. Cotrim P, de Andrade CR, Martelli-Junior H, et al. Expression of matrix metalloproteinases in cyclosporin-treated gingival fibroblasts is regulated by transforming growth factor (TGF)-beta1 autocrine stimulation. J Periodontol. 2002;73(11):1313-1322.
48. Arora P, Silvestri L, Ganss B, et al. Mechanism of cyclosporin-induced inhibition of intracellular collagen degradation. J Biol Chem. 2001;276(17):14100-14109.
49. Rusnak F, Mertz P. Calcineurin: form and function. Physiol Rev. 2000;80(4):1483-1521.
50. Ballard JB, Butler WT. Proteins of the periodontium. Biochemical studies on the collagen and noncollagenous proteins of human gingivae. J Oral Pathol. 1974;3(4):176-184.
51. Dahllöf G, Modéer T, Reinholt FP, et al. Proteoglycans and glycosaminoglycans in phenytoin-induced gingival overgrowth. J Periodontal Res. 1986;21(1):13-21.
52. Henderson JS, Flynn JC, Tucci MA, et al. Site-specific variations in metabolism by human fibroblasts exposed to nifedipine in vitro. J Oral Pathol Med. 1997;26(1):6-10.
53. Modéer T, Andersson G. Regulation of epidermal growth factor receptor metabolism in gingival fibroblasts by phenytoin in vitro . J Oral Pathol Med. 1990;19(4):188-191.
54. Markopoulos AK, Belazi M, Drakoulakos D, et al. Epidermal growth factor in saliva and serum of patients with cyclosporin-induced gingival overgrowth. J Periodontal Res. 2001;36(2):88-91.
55. Bartold PM. Turnover in periodontal connective tissues: dynamic homeostasis of cells, collagen and ground substances. Oral Dis. 1995;1(4):238-253.
56. Carpenter G, Cohen S. Epidermal growth factor. J Biol Chem. 1990;265(14):7709-7712.
57. Buduneli N, Sağol O, Atilla G, et al. Immunohistochemical analysis of epidermal growth factor receptor in cyclosporin A-induced gingival overgrowth. Acta Odontol Scand. 2001;59(6):367-371.
58. Modéer T, Mendez C, Dahllöf G, et al. Effect of phenytoin medication on the metabolism of epidermal growth factor receptor in cultured gingival fibroblasts. J Periodontal Res. 1990;25(2):120-127.
59. Dill RE, Miller EK, Weil T, et al. Phenyotin increases gene expression for platelet-derived growth factor B chain in macrophages and monocytes. J Periodontol. 1993;64(3):169-173.
60. Iacopino AM, Doxey D, Cutler CW, et al. Phenytoin and cyclosporine A specifically regulate macrophage phenotype and expression of platelet-derived growth factor and interleukin-1 in vitro and in vivo: possible molecular mechanism of drug-induced gingival hyperplasia. J Periodontol. 1997;68(1):73-83.
61. Nilsson JM, Sjölund M, Palmberg L, et al. Arterial smooth muscle cells in primary culture produce a platelet-derived growth factor-like protein. Proc Natl Acad Sci U S A. 1985;82(13):4418-4422.
62. Rateitschak-Plüss EM, Hefti A, Lörtscher R, Thiel G. Initial observation that cyclosporin-A induces gingival enlargement in man. J Clin Periodontol. 1983;10(3):237-246.
63. Lucas RM, Howell LP, Wall BA. Nifedipine-induced gingival hyperplasia: A histochemical and ultrastructural study. J Periodontol. 1985;56(4):211-215.